6 Lipoprotein Metabolism and Cholesterol Synthesis
Learning Objectives
- Describe the structure and composition (including associated apoproteins) of the plasma lipoproteins (chylomicrons, VLDL, IDL, LDL, and HDL).
- Compare and contrast the functions of the various lipoprotein particles with respect to their composition, synthesis, transport, and uptake.
- Describe the regulation of cholesterol biosynthesis by the following factors: energy availability, hormones, food intake, and pharmacological manipulation.
- Predict the effect of changes of plasma cholesterol levels on the intracellular synthesis of cholesterol and the transcriptional regulation of genes that are involved in cholesterol homeostasis.
- Predict the consequence of defects in the LDL receptor on plasma lipid levels, receptor number, and the activity of enzymes involved in cellular cholesterol metabolism.
About this Chapter
Cholesterol synthesis and lipoprotein metabolism are several of the most clinically relevant metabolic pathways. Dyslipidemias are a common medical concern, and understanding how to interpret and treat these disorders is fundamental to clinical practice. This section will describe the role, metabolism, and regulation of lipoproteins within circulation.
6.1 Cholesterol Synthesis
Cholesterol is a key component of cell membranes and is an essential precursor for steroid hormone synthesis. All twenty-seven carbons are derived from acetyl-CoA, and the initial synthesis involves the condensation of acetyl-CoA to mevalonate (figure 6.1).
![Cholesterol IUPAC: (3S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol](https://pressbooks.lib.vt.edu/app/uploads/sites/66/2021/11/6.1-scaled.jpg)
Cholesterol synthesis takes place in the cytosol, and the acetyl-CoA needed can be obtained from several sources such as β-oxidation of fatty acids, the oxidation of ketogenic amino acids, such as leucine and lysine, and the pyruvate dehydrogenase reaction (acetyl-CoA shuttled out of the mitochondria is in the form of citrate, which is cleaved into acetyl-CoA and pyruvate by citrate lyase). The process of cholesterol synthesis involves four stages (figure 6.2); however, only the first stage is regulated and will be focused on here.
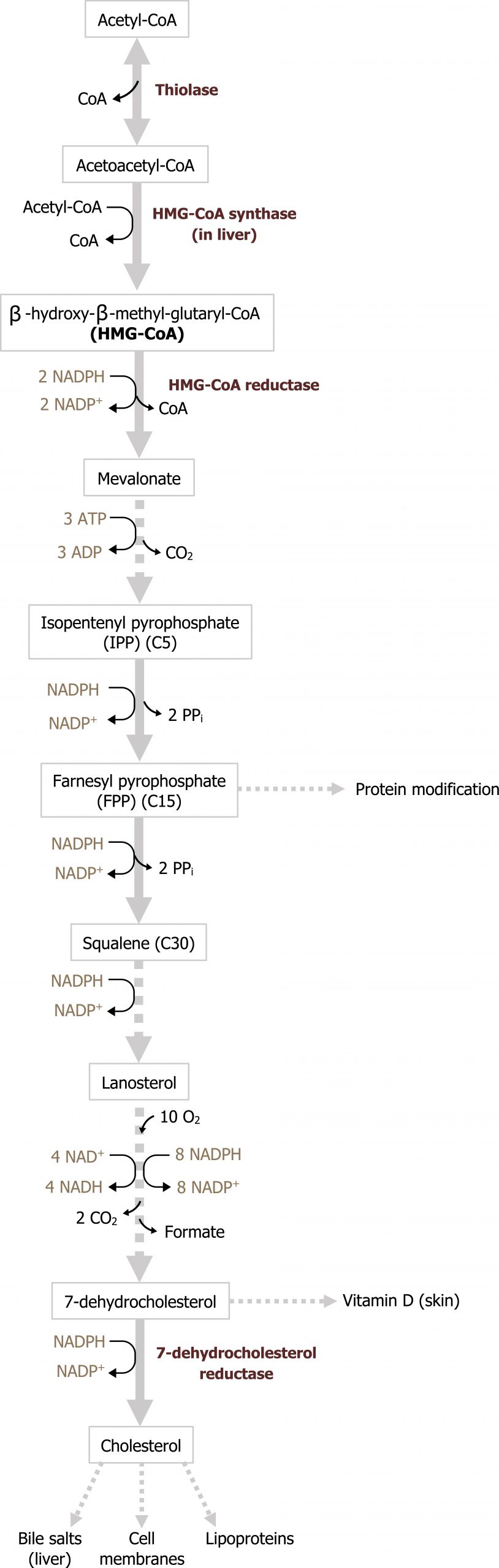
Synthesis of mevalonate from acetyl-CoA
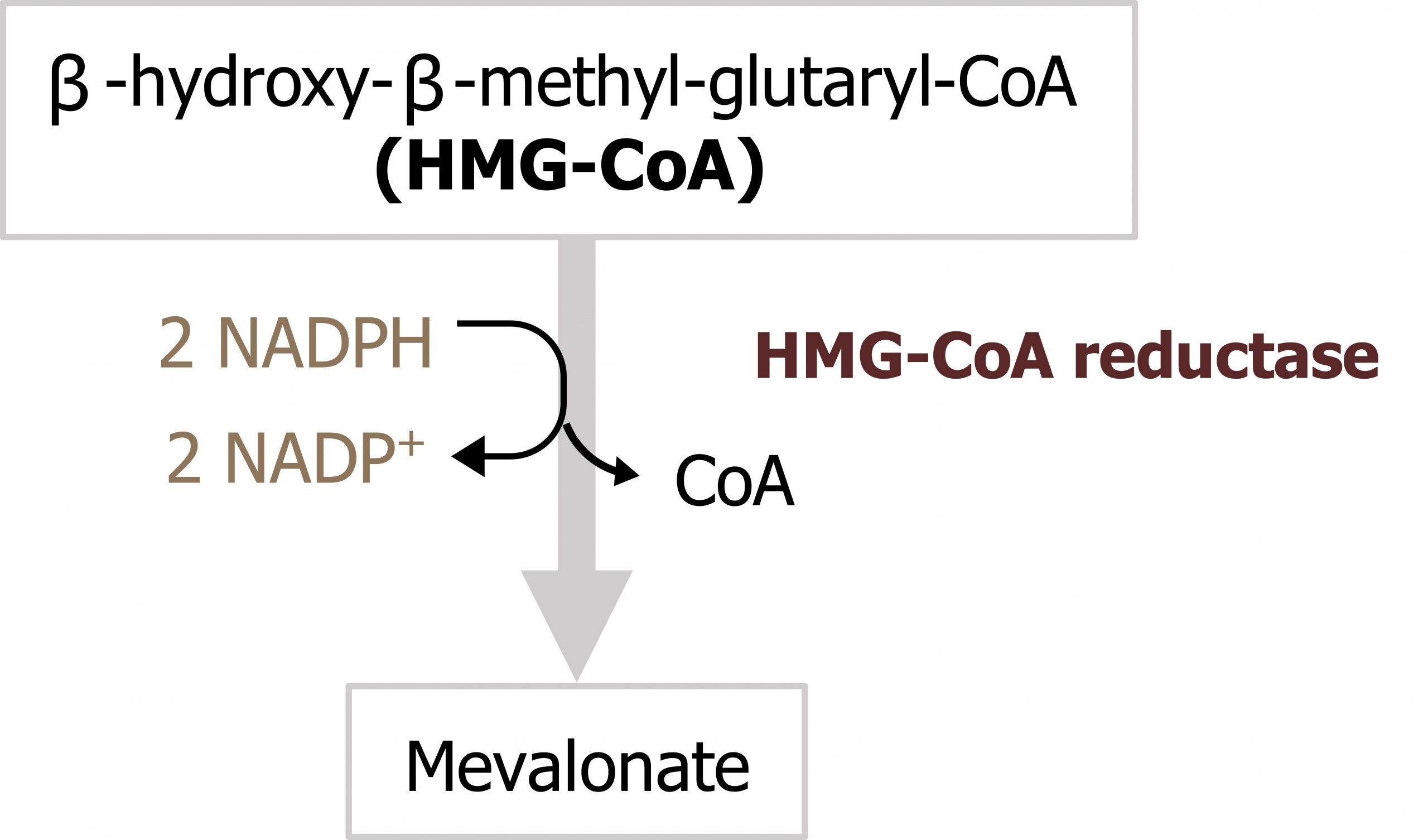
The first stage of cholesterol synthesis leads to the production of the intermediate mevalonate. The synthesis of mevalonate is the committed, rate-limiting step in cholesterol formation. In this reaction, two molecules of acetyl-CoA condense, forming acetoacetyl-CoA, which then condenses with a third molecule of acetyl-CoA to yield the six-carbon compound β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) (figure 6.3) (the cytosolic HMG-CoA synthase in this reaction is distinct from the mitochondrial HMG-CoA synthase that catalyzes a similar reaction involved in production of ketone bodies). The committed step and major point of regulation of cholesterol synthesis involves reduction of HMG-CoA to mevalonate, in a reaction that is catalyzed by HMG-CoA reductase.
The subsequent steps of the pathway proceed largely unregulated, and mevalonate is used to synthesize isoprenoid units (five-carbon units). These five-carbon chains are joined in a head-to-tail fashion generating squalene, thirty-carbons, which undergoes a cyclization reaction after epoxidation. The cyclized product, lanosterol, undergoes several reactions to generate the final product, cholesterol.
Regulation of cholesterol synthesis
The major regulatory enzyme for cholesterol synthesis is HMG-CoA reductase. This enzyme is tightly controlled by many different types of regulation and can be influenced by hormonal changes as well as cellular needs (figure 6.4). This is also one of the primary pharmacological targets for the management of hypercholesterolemia. The statins are direct inhibitors of this enzyme.
Transcriptional control
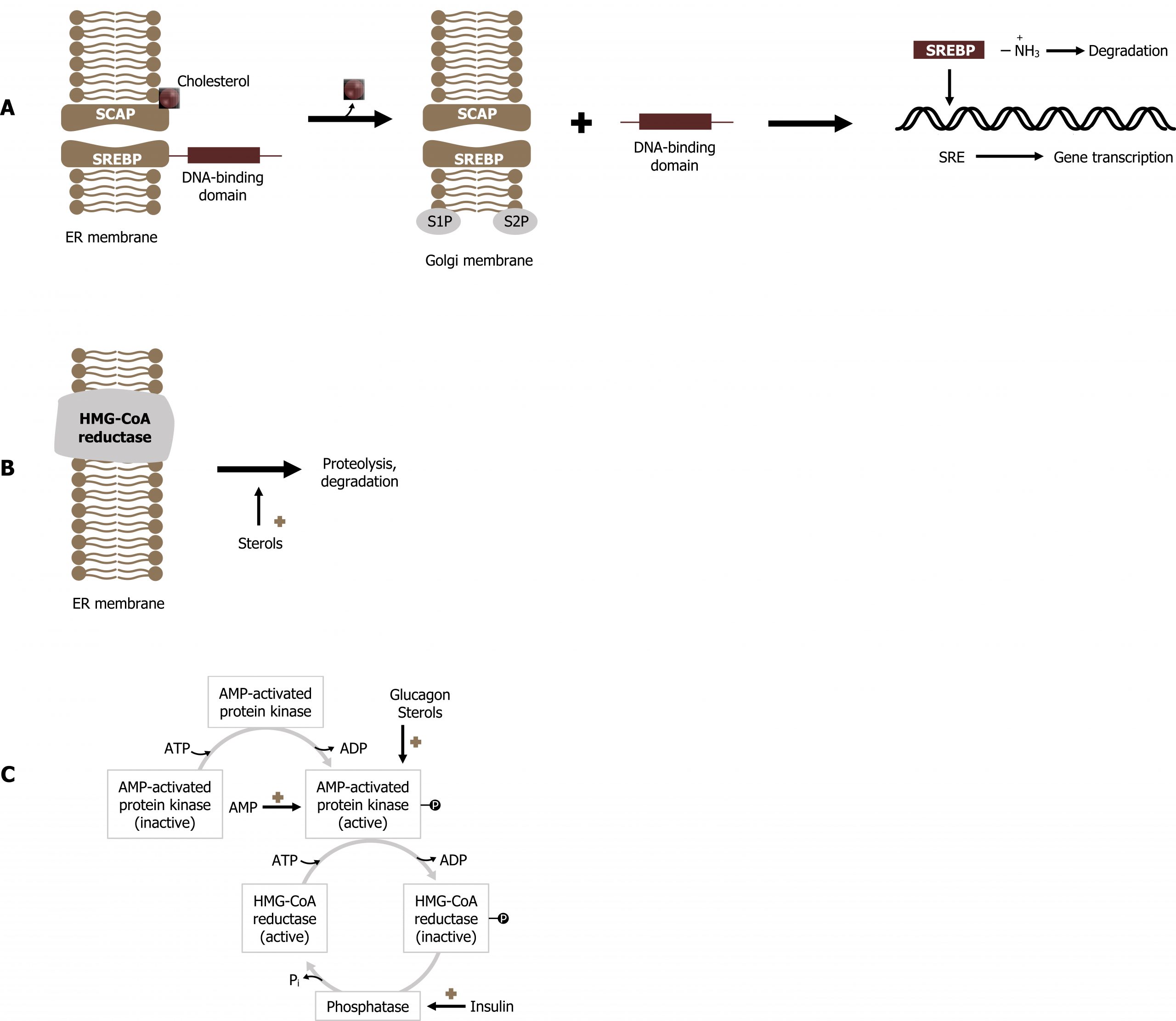
The rate of synthesis of HMG-CoA reductase messenger RNA (mRNA) is controlled by one of the family of sterol-regulatory element-binding proteins (SREBPs). SREBPs are integral proteins of the endoplasmic reticulum (ER). When cholesterol levels in the cell are high, the SREBP is bound to SCAP (SREBP cleavage activating protein) in the ER membrane. When cholesterol levels drop, the sterol leaves its SCAP-binding site, and the SREBP:SCAP complex is transported to the Golgi apparatus. Within the Golgi, two proteolytic cleavages occur, which release the N-terminal transcription factor domain from the Golgi membrane. Once released, the active amino terminal component travels to the nucleus to bind to sterol-regulatory elements (SREs). Binding to this upstream element enhances transcription of the HMG-CoA reductase gene. The soluble SREBPs are rapidly turned over and need to be continuously produced to stimulate reductase mRNA transcription effectively. As cholesterol levels in the cell increase, due to de novo synthesis, cholesterol will bind to SCAP and prevent translocation of the complex to the Golgi, leading to a decrease in transcription of the reductase gene and thus less reductase protein being produced (figure 6.4).
Proteolytic degradation of HMG-CoA reductase
The amount of HMG-CoA reductase can also be influenced by proteolytic degradation. The membrane domains of HMG-CoA reductase contain sterol-sensing regions, which are similar to those in SCAP. As levels of cholesterol (or its derivatives) increase in the cell, this causes a change in the oligomerization state of the membrane domain of HMG-CoA reductase, rendering the enzyme more susceptible to proteolysis. This, in turn, decreases the activity of the enzyme.
Regulation by covalent modification
Much like other anabolic enzymes, the activity of HMG-CoA reductase can be influenced by phosphorylation. Elevated glucagon levels increase phosphorylation of the enzyme, thereby inactivating it, whereas hyperinsulinemia increases the activity of the reductase by activating phosphatases, which dephosphorylate the reductase. Increased levels of intracellular sterols may also increase phosphorylation of HMG-CoA reductase, thereby reducing its activity as well (feedback suppression).
Adenosine monophosphate (AMP)-activated protein kinase can also phosphorylate and inactivate HMG-CoA reductase. Thus, cholesterol synthesis decreases when ATP levels are low and increases when ATP levels are high, similar to what occurs with fatty acid synthesis (recall that acetyl-CoA carboxylase is also phosphorylated and inhibited by the AMP-activated protein kinase; section 4.4.)
Several fates of cholesterol
Almost all mammalian cells are capable of producing cholesterol. Most of the biosynthesis of cholesterol occurs within liver cells, although the gut, the adrenal cortex, and the gonads (as well as the placenta in pregnant women) also produce significant quantities of the sterol. A small portion of hepatic cholesterol is used for the synthesis of hepatic membranes, but the bulk of synthesized cholesterol is secreted from the hepatocyte as one of three compounds: cholesterol esters, biliary cholesterol (cholesterol found in the bile), or bile acids.
Cholesterol esterification and transport
![Lecithin arrow with enzyme LCAT and addition of cholesterol and cholesterol ester to lysolecithin.Lecithin IUPAC ID: 1,3-di(octadecanoyloxy)propan-2-yl 2-(trimethylazaniumyl)ethyl phosphate. Cholesterol ester IUPAC ID: [(3S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl] (5Z,8Z,11Z)-icosa-5,8,11-trienoate. Lysolecithin IUPAC ID: (3-hexadecanoyloxy-2-hydroxypropyl) 2-(trimethylazaniumyl)ethyl phosphate. Cholesterol IUPAC ID: described in figure 6.1.](https://pressbooks.lib.vt.edu/app/uploads/sites/66/2021/11/6.5-1.png)
Cholesterol is an amphipathic molecule (containing both polar and nonpolar regions), and in its native state it can freely diffuse through membranes. In order to be stored in cells, cholesterol must be modified by increasing its hydrophobicity. Cholesterol ester production in the liver is catalyzed by acyl-CoA‒cholesterol acyl transferase (ACAT). ACAT catalyzes the transfer of a fatty acid from coenzyme A to the hydroxyl group on carbon 3 of cholesterol. (This is similar to the reaction catalyzed by lecithin:cholesterol acyltransferase within the plasma associated with HDLs; figure 6.5.) Regardless of whether the additional group is an acyl chain or phosphatidylcholine, the resulting cholesterol esters are more hydrophobic than free cholesterol. The liver packages some of the esterified cholesterol into the hollow core of lipoproteins, primarily VLDL. VLDL is secreted from the hepatocyte into the blood and transports the cholesterol esters (triacylglycerols, phospholipids, apoproteins, etc.) to the tissues that require greater amounts of cholesterol than they can synthesize de novo. These tissues then use the cholesterol for the synthesis of membranes, the formation of steroid hormones, and the biosynthesis of vitamin D.
Synthesis of specialized products
The hepatic cholesterol pool serves as a source of cholesterol for the synthesis of the relatively hydrophilic bile acids and their salts. These derivatives of cholesterol are effective detergents because they contain both polar and nonpolar regions. They are introduced into the biliary ducts of the liver. They are stored and concentrated in the gallbladder and later discharged into the gut in response to the ingestion of food. Finally, cholesterol is the precursor of all five classes of steroid hormones: glucocorticoids, mineralocorticoids, androgens, estrogens, and progestins. Cholesterol and steroid hormones are transported through the blood from their sites of synthesis to their target organs. Because of their hydrophobicity, they must be complexed with a serum protein. Serum albumin can act as a nonspecific carrier for the steroid hormones, but there are specific carriers as well (section 2.1).
6.1 References and resources
Text
Ferrier, D. R., ed. Lippincott Illustrated Reviews: Biochemistry, 7th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2017, Chapter 15: Metabolism of Dietary Lipids, Chapter 18: Cholesterol and Steroid Metabolism.
Le, T., and V. Bhushan. First Aid for the USMLE Step 1, 29th ed. New York: McGraw Hill Education, 2018, 92–94.
Lieberman, M., and A. Peet, eds. Marks’ Basic Medical Biochemistry: A Clinical Approach, 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2018, Chapter 29: Digestion and Transport of Dietary Lipids, Chapter 32: Cholesterol Absorption: Synthesis, Metabolism and Fate Section.
Figures
Grey, Kindred, Figure 6.1 Structure of cholesterol. 2021. Chemical structure by Henry Jakubowski. https://archive.org/details/6.1_20210924. CC BY 4.0.
Grey, Kindred, Figure 6.2 Cholesterol synthetic pathway. 2021. https://archive.org/details/6.2_20210924. CC BY 4.0.
Grey, Kindred, Figure 6.3 Regulatory step catalyzed by HMG-CoA reductase. 2021. https://archive.org/details/6.3_20210924. CC BY 4.0.
Grey, Kindred, Figure 6.5 Esterification of cholesterol by LCAT. 2021. Chemical structure by Henry Jakubowski. https://archive.org/details/6.5_20210924. CC BY 4.0.
Lieberman M, Peet A. Figure 6.4 Regulation of cholesterol synthesis. Adapted under Fair Use from Marks’ Basic Medical Biochemistry. 5th Ed. pp 647. Figure 32.6 Regulation of 3-hydroxymethylglutryl coenzyme A (HMG-CoA reductase activity. 2017. Added squiggle by Made by Made from the Noun Project and ion channel by Léa Lortal from the Noun Project.
6.2 Lipid Transport
Most of the lipids found in the body fall into the categories of fatty acids and triacylglycerols (TAGs); glycerophospholipids and sphingolipids; eicosanoids; cholesterol, bile salts, and steroid hormones; and fat-soluble vitamins. These lipids have very diverse chemical structures and functions. However, they are related by a common property, their relative insolubility in water.
As such, a transport system for distribution of major lipids is in place to aid in the movement of these compounds. This system involves the family of lipoproteins, which have distinct roles in carrying dietary lipids, lipids synthesized through de novo mechanism in the liver, and for reverse cholesterol transport (figure 6.6).

In addition to the lipid components of lipoproteins, they contain protein components termed apoproteins. The complement of apoproteins on each lipoprotein is unique and is a distinguishing characteristic of each family of lipoproteins. The apoproteins (“apo” describes the protein within the shell of the particle in its lipid-free form) not only add to the hydrophilicity and structural stability of the particle, but they have other functions as well: (1) They activate certain enzymes required for normal lipoprotein metabolism, and (2) they act as ligands on the surface of the lipoprotein that target specific receptors on peripheral tissues that require lipoprotein delivery for their innate cellular functions.
Chylomicrons: Transport of dietary lipids
Fatty acids, which are stored as TAGs, serve as fuels, providing the body with its major source of energy. TAGs are the major dietary lipids and are digested in the lumen of the intestine. The initial digestive products, free fatty acids and 2-monoacylglycerol, are reconverted to TAGs in intestinal epithelial cells, packaged in lipoproteins known as chylomicrons, and secreted into the lymph (figure 6.7).
Chylomicrons are the largest lipoproteins and contain cholesterol and fat-soluble vitamins, in addition to their major component, dietary TAGs. The major apoprotein associated with chylomicrons as they leave the intestinal cells is ApoB-48. (The B-48 apoprotein is structurally and genetically related to the B-100 apoprotein synthesized in the liver that serves as a major protein of VLDL.) Microsomal transfer protein (MTP) aids in the loading of apoB-48 protein onto the chylomicron before the nascent chylomicron is secreted. Nascent chylomicrons are secreted by the intestinal epithelial cells into the chyle of the lymphatic system and enter the blood through the thoracic duct. Nascent chylomicrons begin to enter the blood within one to two hours after the start of a meal; as the meal is digested and absorbed, they continue to enter the blood for many hours. Chylomicron maturation occurs in circulation as they accept additional apoproteins from high-density lipoprotein (HDL) (figures 6.7 and 6.10).
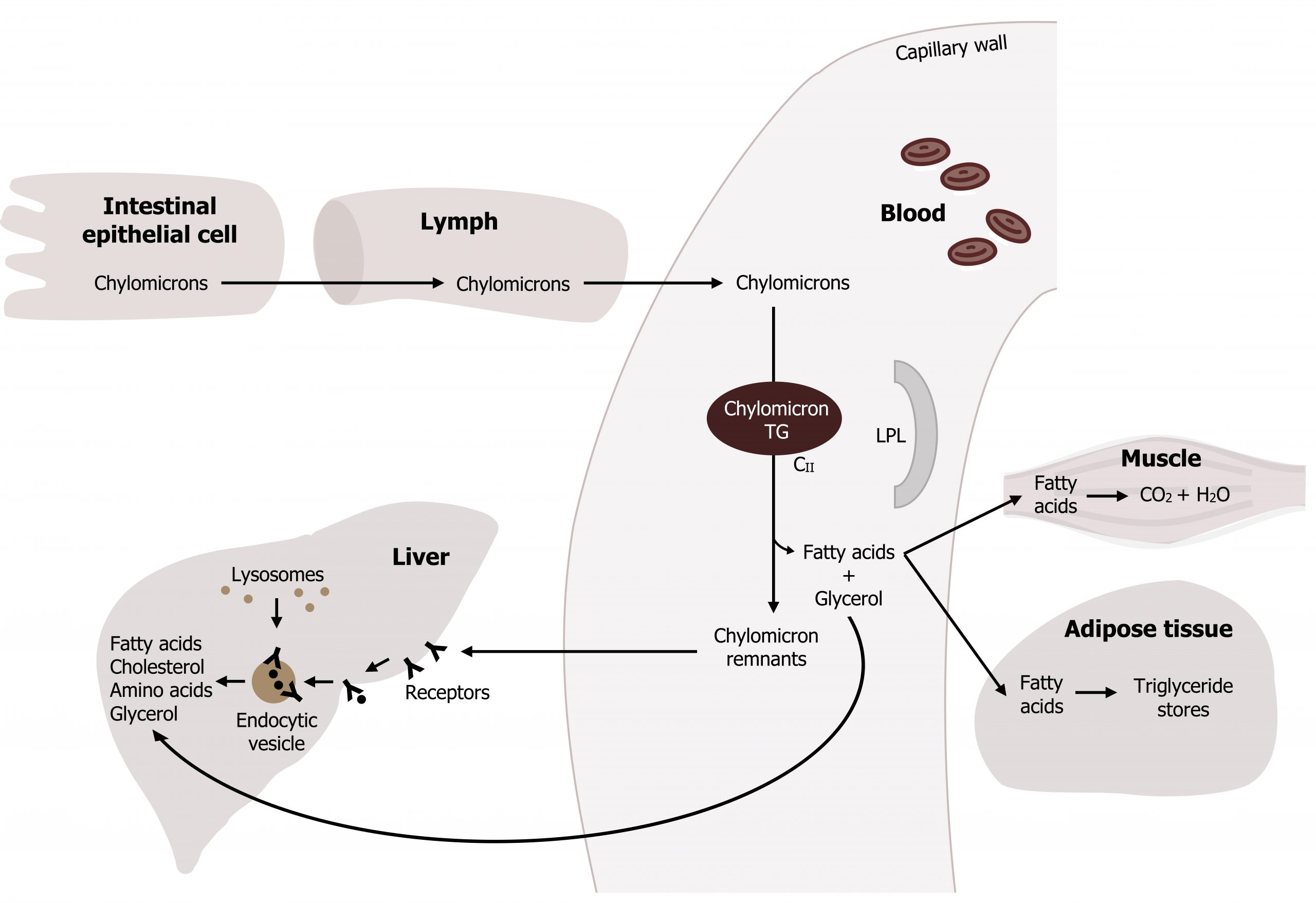
HDL predominantly transfers apoproteins E and CII to the nascent chylomicrons. ApoE is recognized by membrane receptors, and this interaction allows apoE-bearing lipoproteins to enter these cells by endocytosis; once inside the cell the particle is broken down through a lysosomal-mediated process. ApoCII acts as an activator of lipoprotein lipase (LPL), the enzyme on capillary endothelial cells, which digests the TAGs of the chylomicrons and VLDLs in the blood.
Fate of chylomicrons
The TAGs transported by chylomicrons are hydrolyzed by lipoprotein lipase (LPL), an enzyme present on endothelial cells that line the capillary walls. ApoCII on the chylomicron will interact with LPL and activate the enzyme. Insulin stimulates the synthesis and secretion of LPL so that after a meal, when triglyceride levels increase in circulation, LPL is upregulated (through insulin release) to facilitate the hydrolysis of fatty acids from the triglyceride.
Therefore, adipose LPL is more active after a meal, when chylomicron levels are elevated in the blood. The fatty acids released from TAGs by LPL are eventually repackaged in the adipose tissue and stored as TAGs within the tissue.
The portion of a chylomicron that remains in the blood after LPL action is known as a chylomicron remnant. The remnant has returned (or lost) many of the apoC molecules bound to the mature chylomicron, exposing apoE. The remaining remnant binds to apoE receptors on hepatocytes, and is taken up by the process of endocytosis. Lysosomes fuse with the endocytic vesicles, and the chylomicron remnants are degraded by lysosomal enzymes. The products released through this degradation process (e.g., amino acids, fatty acids, cholesterol, etc.) can be recycled within the cell.
VLDL: Transport of TAGs and cholesterol synthesized in the liver
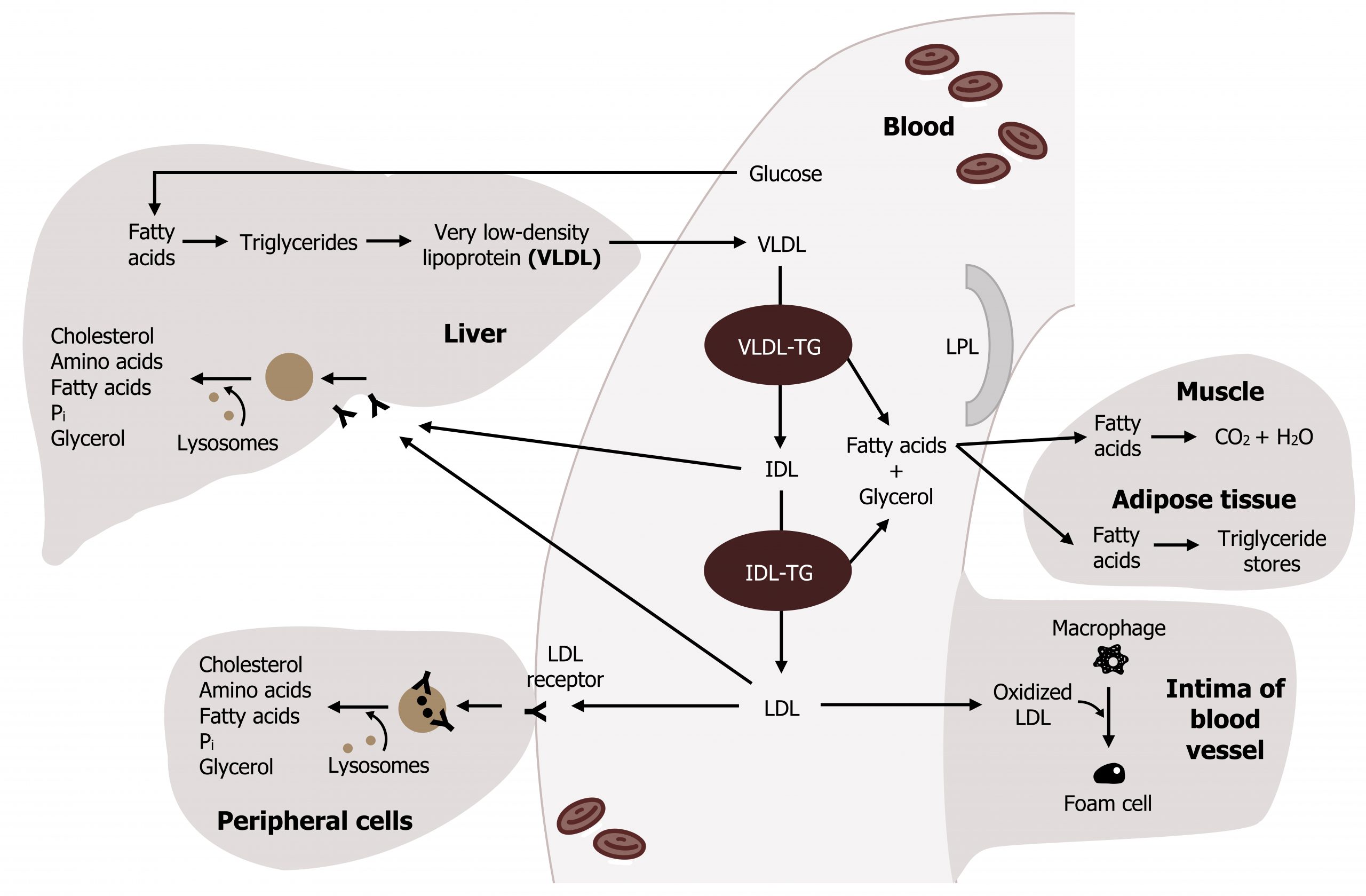
Very low-density lipoprotein (VLDL) is produced in the liver, mainly from lipogenesis. Lipogenesis is an insulin-stimulated process through which excess glucose is converted to fatty acids (section 4.4), which are subsequently esterified to glycerol to form TAGs. TAGs produced in the smooth endoplasmic reticulum of the liver are packaged with cholesterol, phospholipids, and proteins (synthesized in the rough endoplasmic reticulum) to form VLDLs. Apart from their initial origin, VLDLs and chylomicrons are very similar with respect to maturation and activity. The VLDL particles acquire apoB-100 through an MTP-mediated reaction before being released into circulation. Within circulation, VLDLs also interact with HDL and acquire ApoCII and ApoE (figure 6.8). Like chylomicrons, VLDLs are also hydrolyzed by lipoprotein lipase (LPL), and the released fatty acids can be taken up by muscle and other tissues to be oxidized. After a meal, these fatty acids are also taken up by adipose tissue and stored as TAGs. In summary, the process of dietary versus de novo lipid transport has many parallels, which are compared in figure 6.9.
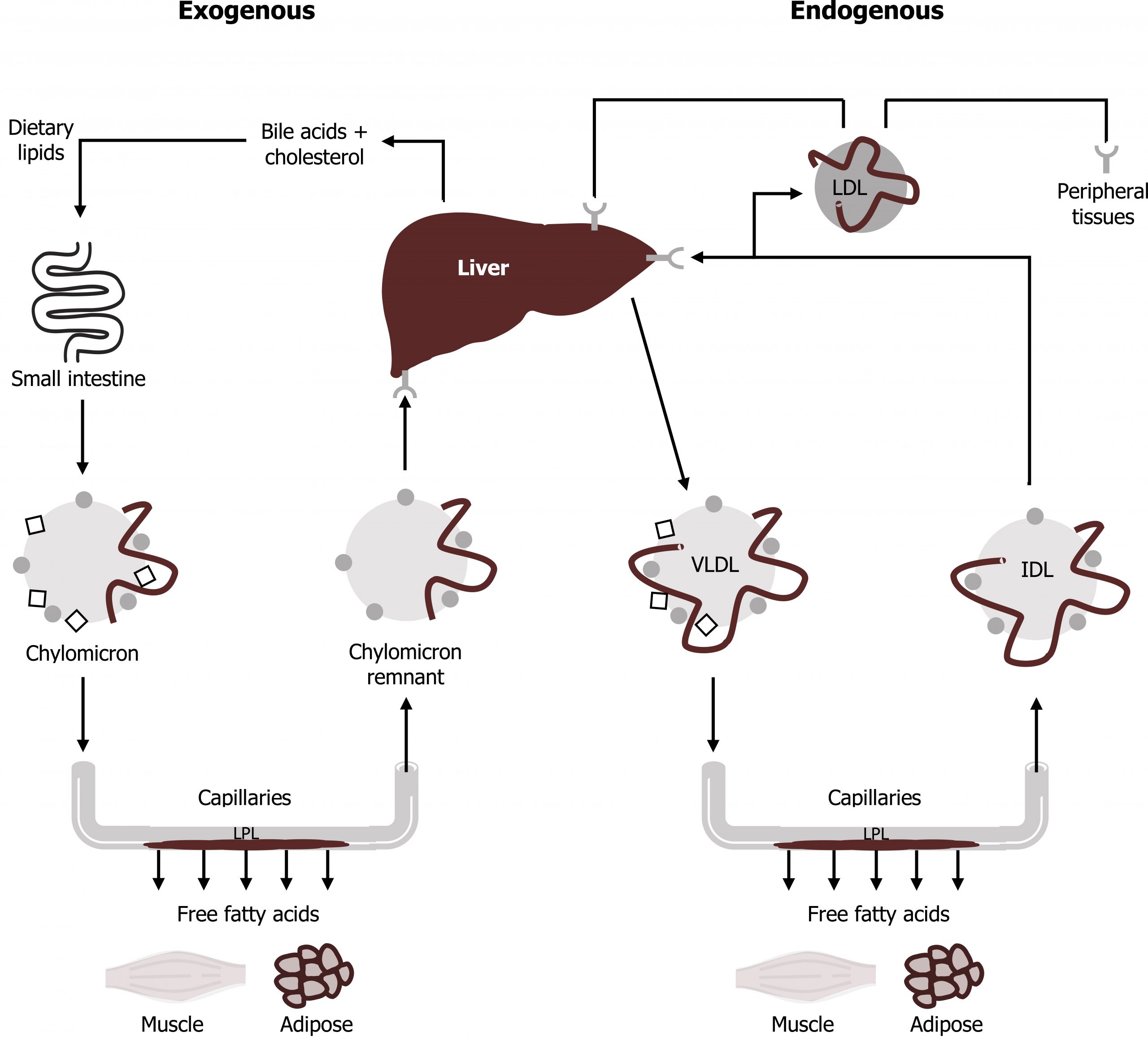
Although VLDLs and chylomicrons have similar roles in the cell, it is important to keep them distinct. The comparison between the transport of exogenous lipids and endogenous lipids is illustrated in figure 6.9. Because the fatty acids stored in adipose tissue come both from chylomicrons and VLDL, we produce our major fat stores both from dietary fat (which is transported by chylomicrons) and dietary sugar (which can be synthesized into TAGs and packaged into VLDL). An excess of dietary protein also can be used to produce the fatty acids for VLDL synthesis. Clinically, measured triacylglycerols (under fasting conditions) will largely reflect the VLDL contribution.
Fate of VLDL
Much like the conversion of chylomicrons to chylomicron remnants, LPL converts VLDL to an intermediate-density lipoprotein (IDL). IDLs, having relatively low TAG content, are taken up by the liver through endocytosis, and degraded lysosomes as discussed above. IDL may also be converted to low-density lipoprotein (LDL) by further digestion of TAGs. Endocytosis of LDL occurs in peripheral tissues (and the liver) and is the major means of cholesterol transport and delivery to peripheral tissues. LDLs taken up by peripheral tissues will help increase the amount of intracellular cholesterol and therefore influence the regulation of HMG-CoA reductase (figure 6.11).
HDL: Reverse cholesterol transport
The primary function of high-density lipoprotein (HDL) is to transport excess cholesterol obtained from peripheral tissues to the liver. HDL also has other roles integral to lipid transport such as exchanging proteins and lipids with chylomicrons and VLDL. HDL particles can be created by several mechanisms, however, nascent HDLs are primarily secreted from the liver and intestine as a relatively small particles whose shell, like that of other lipoproteins, contains phospholipids, free cholesterol, and a variety of apoproteins, specifically apoAI, apoAII, apoCI, and apoCII. Very low levels of triacylglycerols or cholesterol esters are found in the hollow core of this early, or nascent, version of HDL.
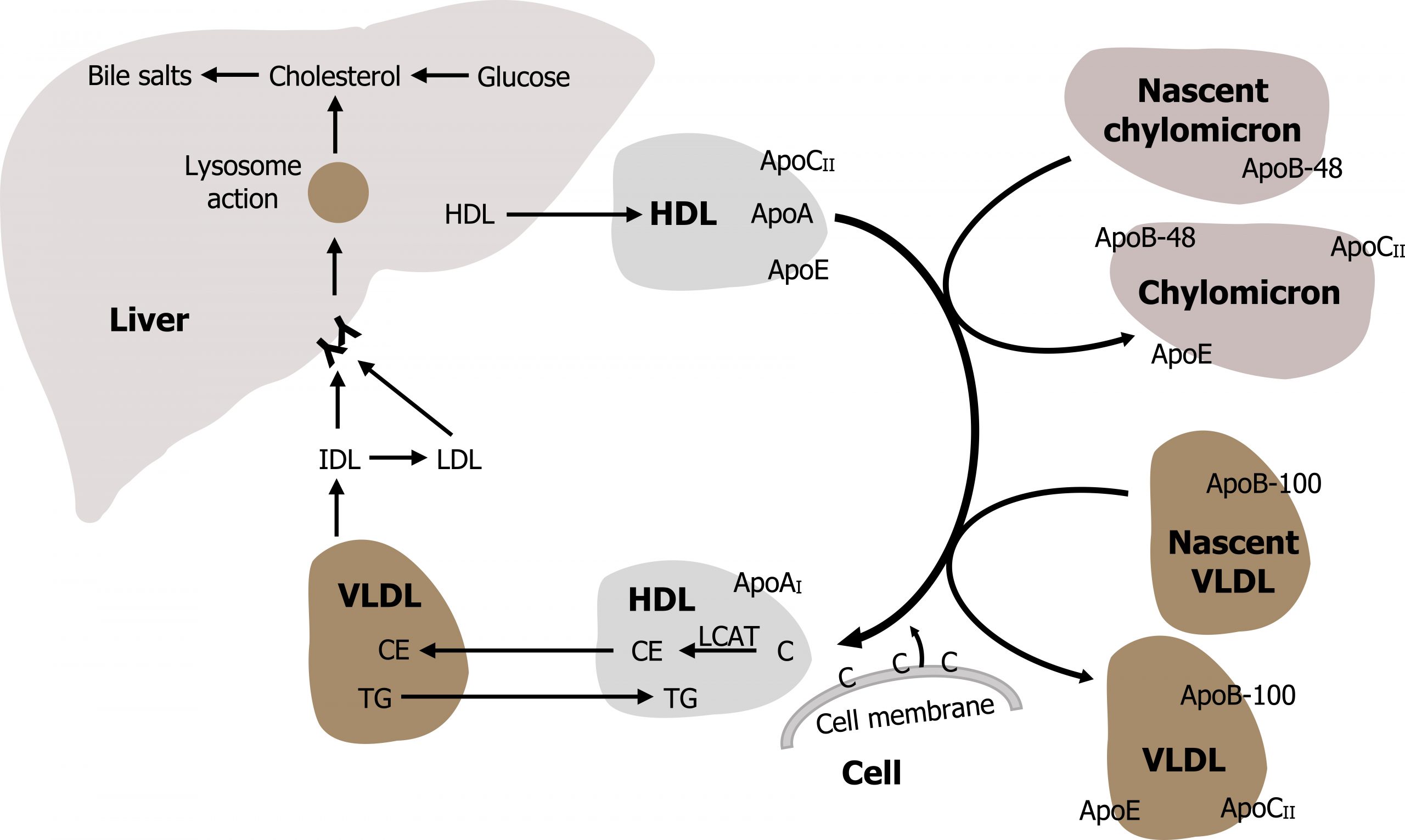
HDLs can also be generated through budding of apoA from chylomicrons and VLDL particles or from free apoAI, which may be shed from other circulating lipoproteins. In this case, the apoAI acquires cholesterol and phospholipids from other lipoproteins and cell membranes, forming a nascent-like HDL particle within the circulation (figure 6.10).
Fate of HDL
In the process of maturation, the nascent HDL particles accumulate phospholipids and cholesterol from cells lining the blood vessels. As the central hollow core of nascent HDL progressively fills with cholesterol esters, HDL takes on a more globular shape to eventually form the mature HDL particle. A major benefit of HDL particles derives from their ability to remove cholesterol from cholesterol-laden cells and to return the cholesterol to the liver, a process known as reverse cholesterol transport. This is particularly beneficial in vascular tissue; by reducing cellular cholesterol levels in the subintimal space, the likelihood that foam cells (lipid-laden macrophages that engulf oxidized LDL cholesterol) will form within the blood vessel wall is reduced.
Reverse cholesterol transport requires a movement of cholesterol from cellular stores to the lipoprotein particle. Cells contain the protein ABCA1 (ATP-binding cassette protein 1) that uses ATP hydrolysis to move cholesterol from the inner leaflet of the membrane to the outer leaflet. Once the cholesterol has reached the outer membrane leaflet, the HDL particle can accept it. To trap the cholesterol within the HDL core, the HDL particle acquires the enzyme lecithin-cholesterol acyltransferase (LCAT) from the circulation (figure 6.10). LCAT catalyzes the transfer of a fatty acid from the 2-position of lecithin (phosphatidylcholine) in the phospholipid shell of the particle to the 3-hydroxyl group of cholesterol, forming a cholesterol ester. The cholesterol esters form the core of the HDL particle and are no longer free to return to the cell.
Mature HDL particles can bind to specific receptors on hepatocytes (such as the apoE receptor), but the primary means of clearance of HDL from the blood is through its uptake by the scavenger receptor SR-B1. This receptor is present on many cell types, and once the HDL particle is bound to the receptor, its cholesterol and cholesterol esters are transferred into the cells. When depleted of cholesterol and its esters, the HDL particle dissociates from the SR-B1 receptor and reenters the circulation.
HDL interactions with other particles
As previously mentioned, HDL plays a key role in the maturation of both chylomicrons and VLDL. First, HDL transfers apoE and apoCII to chylomicrons and to VLDL. The apoCII stimulates the degradation of the TAGs of chylomicrons and VLDL by activating LPL. After digestion of the chylomicrons and the VLDL TAGs, apoE and apoCII are transferred back to HDL.
Another key interaction HDL has with VLDL allows for the redistribution of cholesterol between the two lipoproteins. When HDL obtains free cholesterol from cell membranes, HDL either transports the free cholesterol and cholesterol esters directly to the liver or it can exchange its cholesterol for TAGs in an interaction with VLDL. The cholesterol esterase transfer protein (CETP) resides in circulation and exchanges TAGs from VLDLs with cholesterol-esters from HDL. The greater the concentration of triacylglycerol-rich lipoproteins in the blood, the greater the rate of these exchanges. The CETP exchange pathway may partially explain the observation that whenever triacylglycerol-rich lipoproteins are present in the blood in high concentrations, the amount of cholesterol reaching the liver via cholesterol-enriched VLDL and VLDL remnants increases (figure 6.10), and is consistent with a proportional reduction in the total amount of cholesterol and cholesterol esters that are transferred directly to the liver via HDL.
Lipoprotein receptor-mediated endocytosis
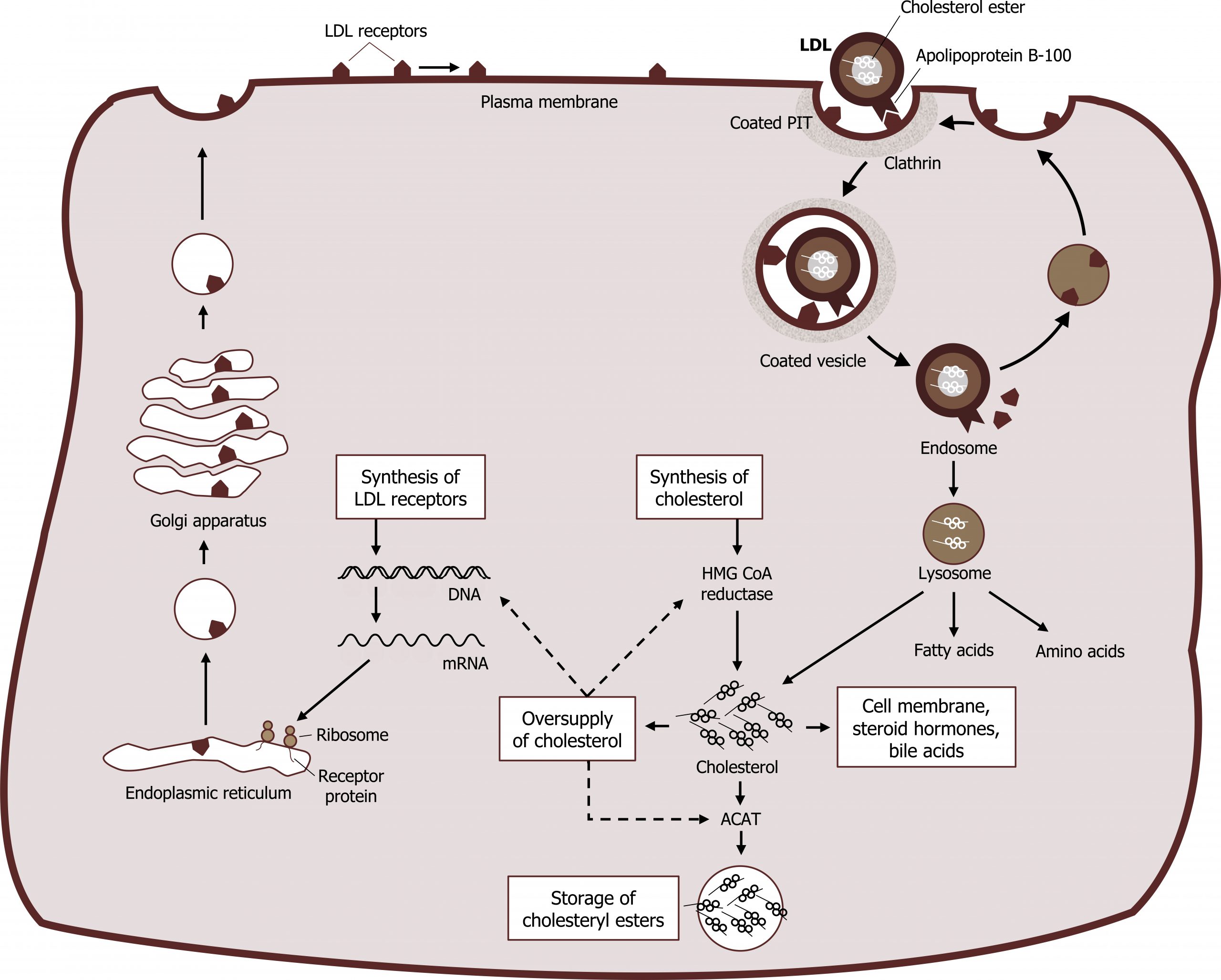
As VLDLs mature to LDLs, these lipoproteins can be taken up through an interaction of the ApoB100 with the LDL receptors on the cell surface. The receptors for LDL are found in clathrin-coated pits within the cell membrane of the target cells. Upon receptor ligand interaction, the plasma membrane in the vicinity of the receptor‒LDL complex invaginates and fuses to form an endocytic vesicle. These vesicles then fuse with lysosomes, and the cholesterol esters of LDL are hydrolyzed to form free cholesterol, which is rapidly re-esterified through the action of ACAT. This rapid re-esterification is necessary to avoid the damaging effect of high levels of free cholesterol on cellular membranes.
The synthesis of the LDL receptor itself is regulated by feedback inhibition as intracellular levels of cholesterol increase. One probable mechanism for this feedback regulation involves one or more of the SREBPs described earlier. These proteins or the cofactors that are required for the full expression of genes that code for the LDL receptor are also capable of sensing the concentration of cholesterol (and its derivatives) within the cell. When sterol levels are high, the process that leads to the binding of the SREBP to the SRE of these genes is suppressed. The rate of synthesis from mRNA for the LDL receptor is reduced under these circumstances. This, in turn, appropriately reduces the amount of cholesterol that can enter these cholesterol-rich cells by receptor-mediated endocytosis (down-regulation of receptor synthesis). When the intracellular levels of cholesterol decrease, these processes are reversed, and cells act to increase their cholesterol levels. Both synthesis of cholesterol from acetyl-CoA and synthesis of LDL receptors are stimulated. An increased number of receptors (up-regulation of receptor synthesis) results in an increased uptake of LDL cholesterol from the blood, with a subsequent reduction of LDL cholesterol levels. At the same time, the cellular cholesterol pool is replenished (figure 6.11).
6.2 References and resources
Text
Ferrier, D. R., ed. Lippincott Illustrated Reviews: Biochemistry, 7th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2017, Chapter 15: Metabolism of Dietary Lipids, Chapter 18: Cholesterol and Steroid Metabolism.
Le, T., and V. Bhushan. First Aid for the USMLE Step 1, 29th ed. New York: McGraw Hill Education, 2018, 92–94.
Lieberman, M., and A. Peet, eds. Marks’ Basic Medical Biochemistry: A Clinical Approach, 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2018, Chapter 29: Digestion and Transport of Dietary Lipids, Chapter 32: Cholesterol Absorption: Synthesis, Metabolism and Fate Section.
Figures
Ferrier D. Figure 6.6 Overview of lipoprotein size and structure. Adapted under Fair Use from Lippincott Illustrated Reviews Biochemistry. 7th Ed. pp 227. Figure 18.13 Plasma lipoprotein particles exhibit a range of sizes and densities, and typical values are shown. 2017.
Ferrier D. Figure 6.11 Uptake of LDL and regulation of cholesterol synthesis. Adapted under Fair Use from Lippincott Illustrated Reviews Biochemistry. 7th Ed. pp 233. Figure 18.20 Cellular uptake and degradation of low-density lipoprotein (LDL) particles. 2017. Added squiggle by Made by Made from the Noun Project.
Lieberman M, Peet A. Figure 6.7 Transport of dietary lipids via chylomicrons. Adapted under Fair Use from Marks’ Basic Medical Biochemistry. 5th Ed. pp 601. Figure 29.11 Fate of chylomicrons. 2017. Added Liver by Liam Mitchell from the Noun Project, Muscle by Laymik from the Noun Project, and red blood cells by Lucas Helle from the Noun Project.
Lieberman M, Peet A. Figure 6.8 Transport of TAGs from de novo synthesis using VLDL. Adapted under Fair Use from Marks’ Basic Medical Biochemistry. 5th Ed. pp 680. Figure 32.12 Fate of very-low-desnity lipoprteins (VLDL). 2017. Added macrophage by Léa Lortal from the Noun Project, Liver by Liam Mitchell from the Noun Project, and red blood cells by Lucas Helle from the Noun Project.
Lieberman M, Peet A. Figure 6.10 Interaction of chylomicrons and VLDL with HDL in circulation. Adapted under Fair Use from Marks’ Basic Medical Biochemistry. 5th Ed. pp 683. Figure 32.15 Functions and fate of high-density lipoprotein (HDL). 2017. Added Liver by Liam Mitchell from the Noun Project.
Loscalzo J. Figure 6.9 Comparison of the role of chylomicrons and VLDLs in lipid transport. Adapted under Fair Use from Harrison’s Cardiovascular Medicine 2 ed. online. Figure 31.2 The exogenous and endogenous lipoprotein metabolic pathways. 2013. Added Small Intestine by PJ Witt from the Noun Project, Liver by Liam Mitchell from the Noun Project, and Muscle by Laymik from the Noun Project.
