8 Amino Acid Metabolism and Heritable Disorders of Degradation
Learning Objectives
- Define essential, conditionally essential, and nonessential amino acids, and explain how certain nonessential amino acids become essential in certain conditions.
- Integrate amino acid synthesis with specific precursors from glycolysis, citric acid cycle, and the pentose phosphate pathway.
- Identify key roles of amino acids as substrates for the synthesis of specialized products including heme, GABA, carnitine, glutathione, serotonin, histamine, ubiquinone, melanin, creatine, and dopamine.
- Review the role of transamination in the interconversion of amino acids and connection to the urea cycle; see section 5.3.
- Distinguish the following disease states associated with inborn errors of metabolism, including (A) deficient enzyme, (B) inheritance pattern of the disease, and (C) relation of the deficiency to the buildup of secondary metabolites.
- The following is a list of diseases to focus on:
- Phenylketonuria (phenylalanine metabolism),
- Homocystinuria (methionine metabolism),
- Maple syrup urine disease (metabolism of branched-chain amino acids), and
- Alkaptouria (tyrosine metabolism).
About this Chapter
There are twenty amino acids required for metabolic homeostasis. Of the twenty amino acids, eleven are considered nonessential, meaning they can be synthesized by the body. With the exceptions of tyrosine and cysteine, the others can be synthesized from glucose and a nitrogen donor. The other nine amino acids are essential and must be supplied by the diet. As mentioned previously, in addition to supplying carbon for gluconeogenesis, amino acids play important roles in the synthesis of essential cellular components. Disruptions of many of these pathways can lead to clinical disorders, many of which are identified during newborn screenings. The synthesis of all amino acids will not be addressed in this section; rather the most clinically relevant pathways will be focused on.
8.1 Amino Acid Metabolism and Specialized Products
Cofactors essential for amino acid metabolism
The metabolism of many amino acids largely relies on the availability of the cofactors pyridoxal phosphate (vitamin B6 or PLP), tetrahydrobiopterin (BH4), and tetrahydrofolate (TH4). It is important to recognize that deficiencies in these cofactors could present in a similar manner as enzymatic deficiencies of specific pathways. Refer to chapter 2 for more information regarding vitamin B6 and folate.
Pyridoxal phosphate (B6 or PLP)
All transamination reactions require PLP as a cofactor. These reactions are essential for moving (or donating) a nitrogen from an amino acid to a keto-acid to generate a different amino acid.
Tetrahydrobiopterin (BH4)
This is a cofactor synthesized from GTP. It is oxidized during hydroxylation reactions, most notably the conversion of phenylalanine to tyrosine. Enzymatic deficiencies leading to decreased synthesis of BH4 can present similar to deficiencies in phenylalanine metabolism.
Tetrahydrofolate (FH4)
Folate can exist in many forms and is often referred to as tetrahydrofolate. FH4 is often found in various forms with a one-carbon group attached. These one-carbon groups, which make up the one-carbon pool, can be oxidized or reduced. One-carbon groups can be transferred to other compounds and play essential roles in the synthesis of glycine from serine, the synthesis of the base thymine (required for DNA synthesis), the purine bases required for both DNA and RNA synthesis, and the transfer of methyl groups to vitamin B12.
Synthesis of specialized products
The following highlights some of the key aspects of amino acid metabolism.
Phenylalanine and tyrosine
Phenylalanine is an essential amino acid, and hydroxylation of Phe by phenylalanine hydroxylase (PAH) generates tyrosine (figure 8.1). This conversion requires BH4, and deficiencies in either the cofactor or the enzyme PAH can result in phenylketonuria. Additionally, the inability to synthesize tyrosine will make this a conditionally essential amino acid and potentially negatively impact the synthesis of downstream compounds illustrated in figure 8.1.
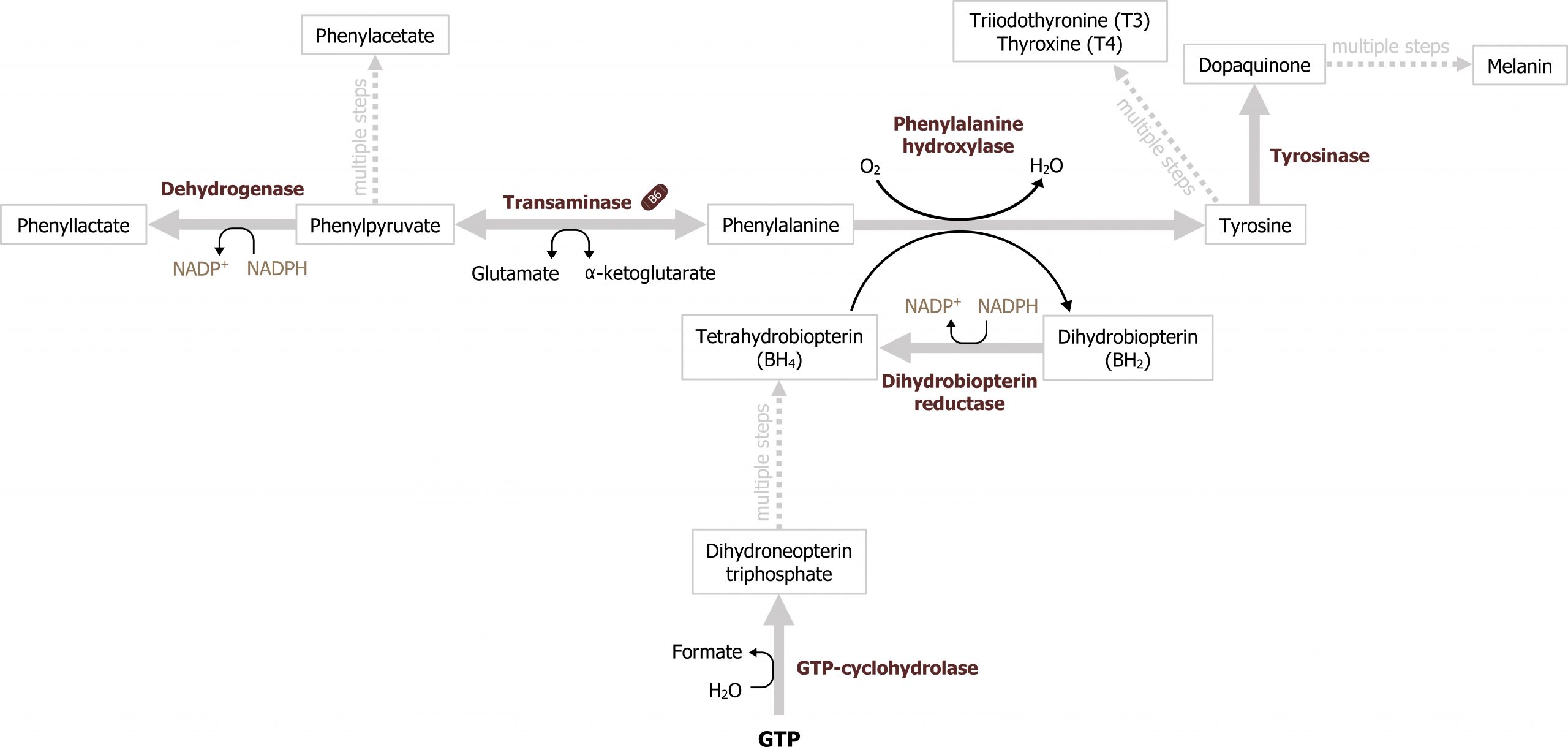
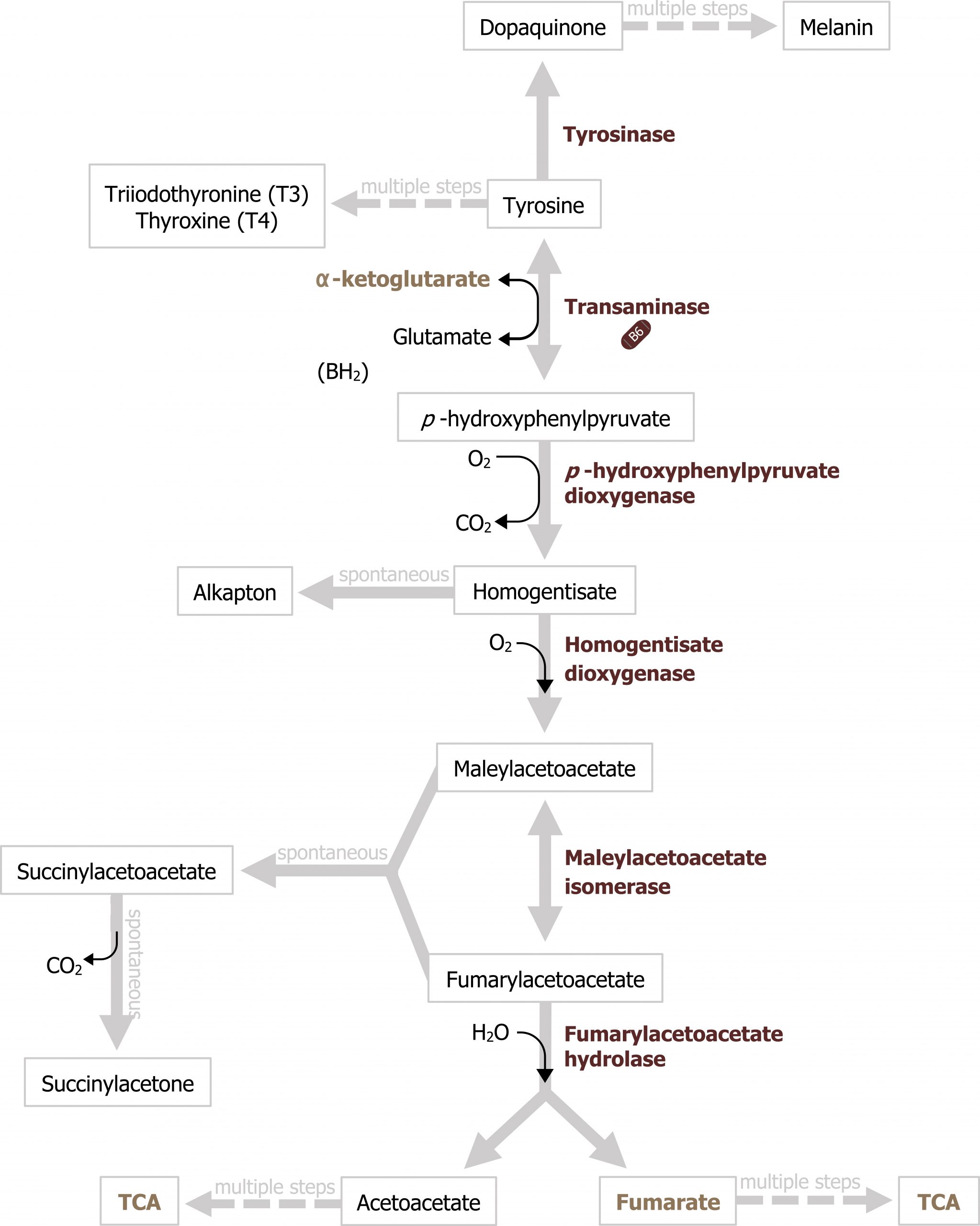
Tyrosine can be produced from phenylalanine metabolism and is required for the production of melanin and the catecholamines. Deficiencies can occur at several different locations in the pathway and result in albinism (tyrosinase), alkaptonuria (homogentisate oxidase), or tyrosinemia, which can manifest due to deficiencies in several enzymes along the pathway (figure 8.2).
Phenylketonuria
Phenylketonuria (PKU) is one of the more common amino acid metabolic disorders and is inherited in an autosomal recessive fashion. There are no symptoms of untreated phenylketonuria in the first months of life, therefore newborn screening is essential for diagnosis and initiation of treatment, which prevents the devastating effects of infantile hyperphenylalaninemia. The screening method detects elevated titers of the amino acid phenylalanine (Phe) in the blood. A positive test result (Phe greater than 150 μmol/L) prompts the physician to begin a phenylalaninerestricted formula and requires a confirmatory quantitative Phe level.
Glycine
Glycine is a key compound that functions as an essential substrate for various pathways including the folate cycle, nucleotide synthesis, and synthesis of porphyrins (heme), glutathione, and creatine. Glycine can be synthesized at the cellular level from 3-phosphoglycerate, an intermediate of glycolysis.
Arginine
Arginine is a nonessential amino acid as it can be produced in the urea cycle. Deficiencies in the urea cycle can cause arginine to become conditionally essential. In these cases, management and supplementation are needed.
Tryptophan
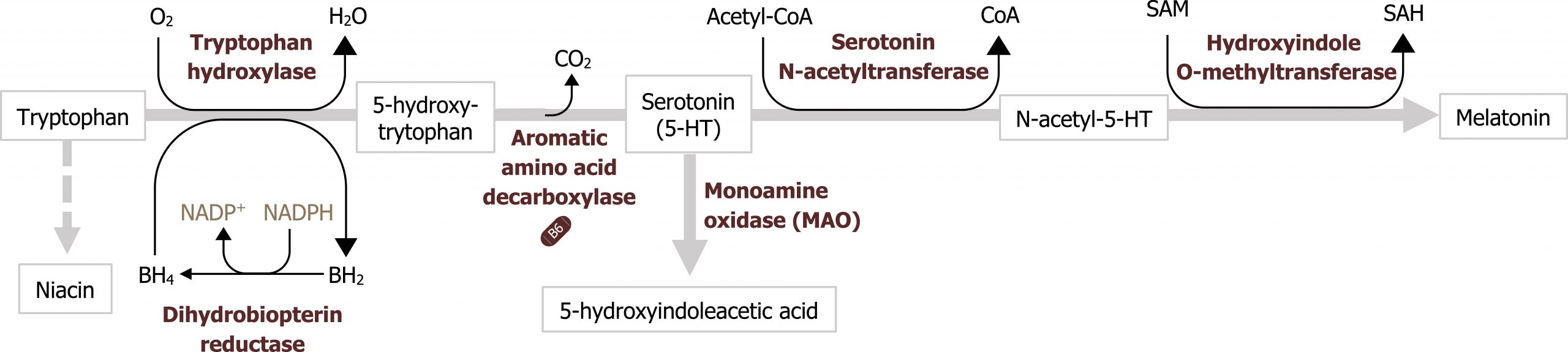
Tryptophan is an essential amino acid that is both ketogenic and glucogenic as it can be oxidized to produce alanine and acetyl-CoA. The ring structure can also be used to synthesize niacin, reducing the dietary requirement for this vitamin. Tryptophan metabolism to serotonin (and subsequently melatonin) requires BH4. Deficiencies here can lead to imbalances in these neurotransmitters (figure 8.3).
Glutamate
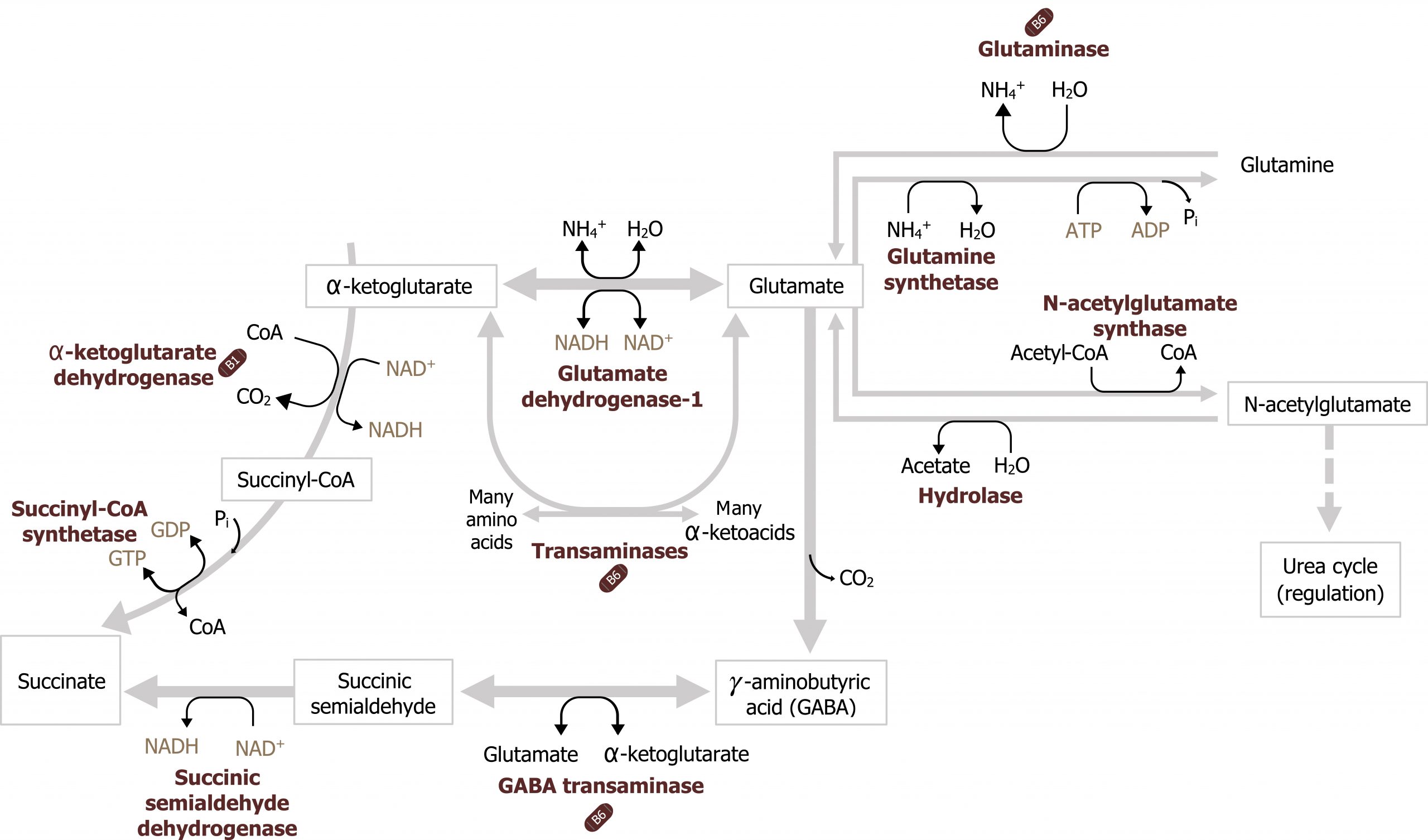
Glutamate plays many key roles in amino acid metabolism and provides substrates for GABA and glutathione synthesis (figure 8.4). Additionally, glutamate plays a key role in nitrogen movement within the body. Glutamate can be deaminated by glutamate dehydrogenase to yield α-ketoglutarate. This can enter directly into the TCA cycle or be transaminated (figure 8.4). Additionally, glutamate can be used to fix or free ammonium to generate glutamine — one of the essential, nontoxic carriers of ammonia.
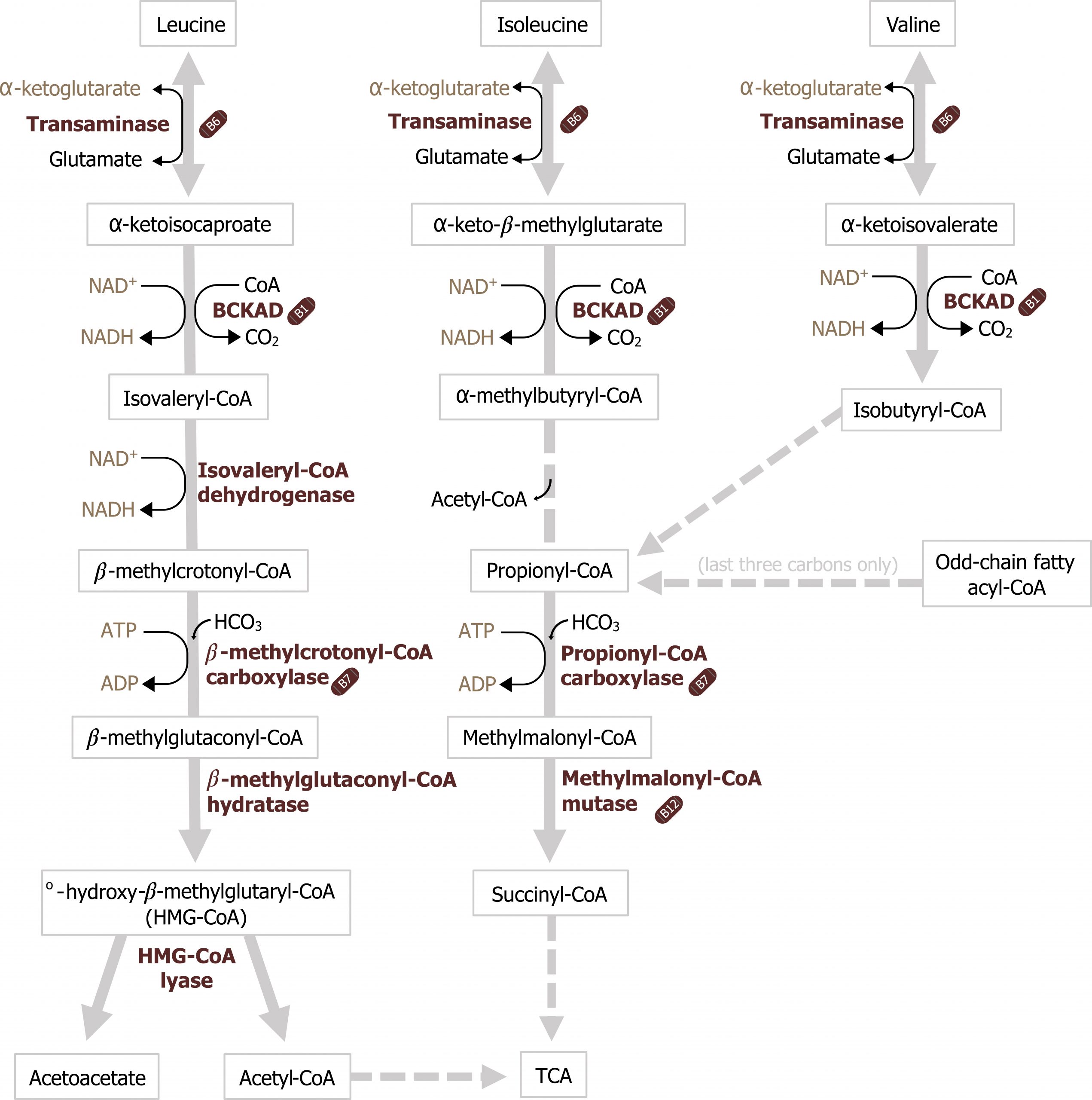
Isoleucine, leucine, and valine (branched-chain amino acids)
Oxidation of these amino acids, collectively described as branched-chain amino acids, occurs in all tissues (except the liver) and is a key fuel source for skeletal muscle. As these amino acids are approximately 25 percent of the amino acid pool, they provide both energy and available substrate to replenish the TCA cycle. The initial step in their metabolism, like all amino acids, is the transamination to generate a keto-acid. These compounds then undergo oxidative decarboxylation by a multiunit enzyme similar to the pyruvate dehydrogenase complex with similar cofactor requirements (section 4.1), and the remaining carbons can enter the TCA cycle.
Maple syrup urine disease
Deficiencies in metabolism of branched-chain amino acids can result in the diagnosis of maple syrup urine disease (MSUD). With an incidence of 1 in 100,000, MSUD is rare even among the inborn errors of metabolism. However, the distinct sweet odor, similar to that of maple syrup, distinguishes this condition as one of the more recognizable metabolic disorders. It is caused by deficient oxidative decarboxylation of α-keto-acid metabolites of leucine, isoleucine, and valine. Affected infants can become symptomatic during the first days of life, with poor feeding, lethargy, seizures, and occasionally coma. Milder forms of MSUD may present later in life, with developmental delays and intellectual disability. Maple syrup urine disease is primarily treated by diet but also by avoiding circumstances that increase catabolism such as high fever and dehydration. If a metabolic crisis occurs, emergency treatment in a hospital is necessary to stabilize the patient.
Methionine
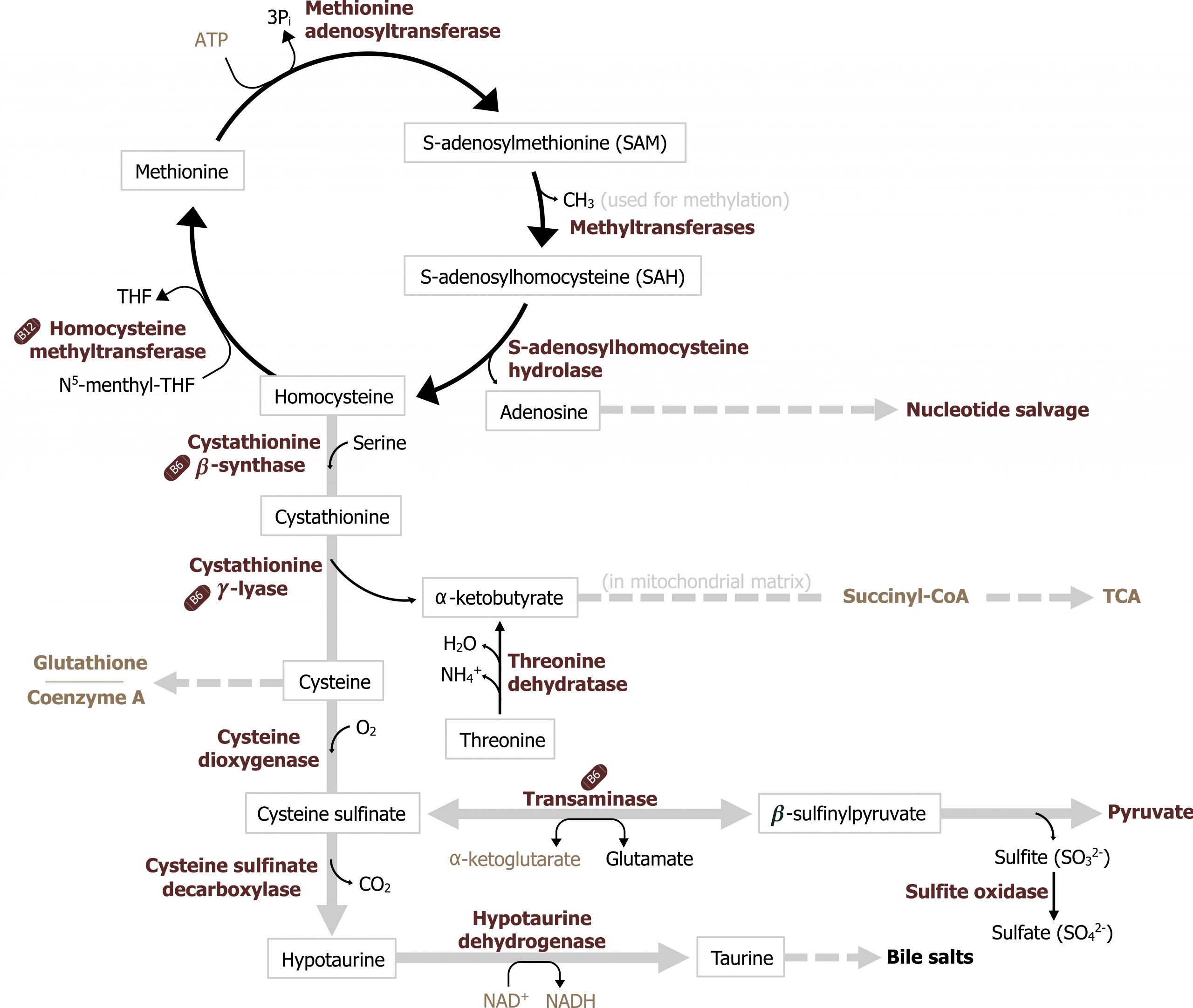
Methionine is an essential amino acid with a complex metabolism of clinical importance. Its metabolism interfaces with the folate cycle, cobalamin remethylation, and the synthesis of S-adenosylmethionine (SAM). Enzymatic or cofactor deficiencies can result in elevated homocysteine levels (hyperhomocysteinemia), which can have negative impacts systemically. Methionine, required for the synthesis of SAM, can be obtained from the diet or produced from remethylation of homocysteine using vitamin B12.
Initially, methionine will condense with ATP to form SAM. SAM has a charged methyl group, which can be transferred to many different acceptor molecules; this step is considered irreversible as the amount of energy released is substantial. SAM is used by many biological pathways to donate methyl groups, and it is in consistent demand. After SAM loses its methyl group, the resulting compound, S-adenosylhomocysteine (SAH), is hydrolyzed to homocysteine and adenosine.
Homocysteine, generated from this reaction, can either be remethylated in a reaction using both folate and cobalamin to resynthesize methionine or can be used for the synthesis or cysteine (figure 8.6).
Remethylation of homocysteine
Homocysteine can be converted back into methionine by using both methyl-FH4 and vitamin B12. (This is the only reaction in which methyl-FH4 can donate the methyl group.) In this reaction, the methyl group from FH4 is transferred to cobalamin associated with homocysteine methyltransferase. Homocysteine receives the methyl group from this charged cobalamin cofactor, and methionine is regenerated. If homocysteine methyltransferase is defective, or if vitamin B12 or FH4 levels are insufficient, homocysteine will accumulate. Elevated homocysteine levels have been linked to cardiovascular and neurological diseases. A consequence of vitamin B12 deficiency is the accumulation of methyl-FH4 and a decrease in other folate derivatives. This is known as the methyl-trap hypothesis; because of the B12 deficiency, most of the carbons in the FH4 pool are trapped in the methyl-FH4 form, which is the most stable. The carbons cannot be released from the folate because the one reaction in which they participate cannot occur because of the B12 deficiency. This leads to a functional folate deficiency, even though total levels of folate are normal.
A folate deficiency (whether functional or actual) leads to megaloblastic anemia caused by an inability of blood cell precursors to synthesize DNA and, therefore, to divide. This leads to large, partially replicated cells being released into the blood to attempt to replenish the cells that have died. Folate deficiencies also have been linked to an increased incidence of neural tube defects, such as spina bifida, in mothers who become pregnant while folate deficient.
Transsulfuration pathway
Further metabolism of homocysteine provides the sulfur atom for the synthesis of cysteine. In this two-step process, homocysteine first reacts with serine to form cystathionine. This is followed by cleavage of cystathionine, which yields cysteine and α-ketobutyrate. The first reaction in this sequence, catalyzed by cystathionine β-synthase, is inhibited by cysteine. Thus, methionine, via homocysteine, is not used for cysteine synthesis unless the levels of cysteine in the body are lower than required for its metabolic functions. An adequate dietary supply of cysteine, therefore, can “spare” (or reduce) the dietary requirement for methionine (figure 8.6).
Consequences of elevated homocysteine
Homocysteine levels can accumulate in several ways, which are related to both folic acid and vitamin B12 metabolism. As SAM is constantly being used as a methyl donor, this results in a consistent production of SAH. Consequently, this leads to constant production of homocysteine. The homocysteine produced can be either remethylated to methionine or condensed with serine to form cystathionine. The major pathway of homocysteine metabolism is remethylation by N5-methyl-FH4, which requires vitamin B12. The liver also contains a second pathway in which betaine (a degradation product of choline) can donate a methyl group to homocysteine to form methionine, but this is a minor pathway. The conversion of homocysteine to cystathionine requires pyridoxal phosphate (PLP). Thus, if an individual is deficient in vitamin B12, the conversion of homocysteine to methionine by the major route is inhibited. This directs homocysteine to produce cystathionine, which eventually produces cysteine. Homocysteine also accumulates in the blood if a mutation is present in the enzyme that converts N5,N10-methylene-FH4 to N5-methyl-FH4. When this occurs, the levels of N5-methyl-FH4 are too low to allow homocysteine to be converted to methionine. The loss of this pathway, coupled with the feedback inhibition by cysteine on cystathionine formation, also leads to elevated homocysteine levels in the blood. A third way in which serum homocysteine levels can be elevated is by a mutated cystathionine β-synthase or a deficiency in vitamin B6, the required cofactor for that enzyme. These defects block the ability of homocysteine to be converted to cystathionine, and the homocysteine that does accumulate cannot all be accommodated by conversion to methionine. Thus, an accumulation of homocysteine results.
8.1 References and resources
Text
Ferrier, D. R., ed. Lippincott Illustrated Reviews: Biochemistry, 7th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2017, Chapter 20: Amino Acid Degradation and Synthesis, Chapter 21: Conversion of Amino Acids to Specialized Products.
Le, T., and V. Bhushan. First Aid for the USMLE Step 1, 29th ed. New York: McGraw Hill Education, 2018, 69, 83–85.
Lieberman, M., and A. Peet, eds. Marks’ Basic Medical Biochemistry: A Clinical Approach, 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2018, Chapter 37: Synthesis and Degradation of Amino Acids, Chapter 39: Tetrahydrofolate, Vitamin B12, and S-Adenosylmethionine.
Figures
Grey, Kindred, Figure 8.1 Metabolism of phenylalanine requires BH4 and also produces tyrosine. Deficiencies in cofactor or phenylalanine hydroxylase can result in phenylketonuria. 2021. https://archive.org/details/8.1-new. CC BY 4.0.
Grey, Kindred, Figure 8.2 Tyrosine can be produced from phenylalanine metabolism and is required for the production of melanin and the catecholamines. Deficiencies can occur at several different locations in the pathway and result in albinism, alkaptonuria or tyrosinemia. 2021. https://archive.org/details/8.2_20210926. CC BY 4.0.
Grey, Kindred, Figure 8.3 Metabolism of tryptophan to melatonin. 2021. https://archive.org/details/8.3_20210926. CC BY 4.0.
Grey, Kindred, Figure 8.4 Glutamate metabolism as it interfaces with nitrogen transport and synthesis of GABA. 2021. https://archive.org/details/8.4_20210926. CC BY 4.0.
Grey, Kindred, Figure 8.5 Metabolism of branched chain amino acids. Deficiencies in BCKAD can result in the presentation of Maple Syrup Urine Disease. 2021. https://archive.org/details/8.5_20210926. CC BY 4.0.
Grey, Kindred, Figure 8.6 Metabolism of methionine. Remethylation and transsulfuration of homocysteine are illustrated. Cofactor or enzymatic deficiencies can result in an elevation of homocysteine. 2021. https://archive.org/details/8.6_20210926. CC BY 4.0.
