4 The Restrictive Lung Diseases
Learning objectives
- Relate the signs and symptoms of an interstitial lung disease (ILD) patient to the pathological process of the disease.
- Differentiate between forms of interstitial lung disease via histological appearance and patient history.
Basis of Restrictive Lung Disease
The restrictive lung diseases are characterized by pathophysiological disruption of the lung interstitial tissue that causes problems with lung expansion. As such, these diseases are more specifically referred to as the interstitial lung diseases, or ILDs. There are about 150 conditions that disrupt lung structure and generally produce a restrictive, rather than obstructive, disorder.
This section provides a broad overview and generalized mechanisms of interstitial lung disease. Subclassifications and details of specific conditions are presented in the next section.
Before we start taking about diseases affecting the lung interstitium, let us remind ourselves of what it is.
The interstitial tissue, sometimes referred to as parenchyma, surrounds the alveolar and capillary structures and contributes to the mechanical behavior of the lungs. The interstitium is extremely thin between the alveoli and capillaries, and forms the basement membrane through which gas exchange occurs. On the parenchymal side of the capillaries the interstitium is more substantial and is more involved in fluid exchange. There is also substantial amounts of interstitial tissue in the spaces around major vessels and airways, and it also makes up the interlobular septa.
Mechanisms of ILD
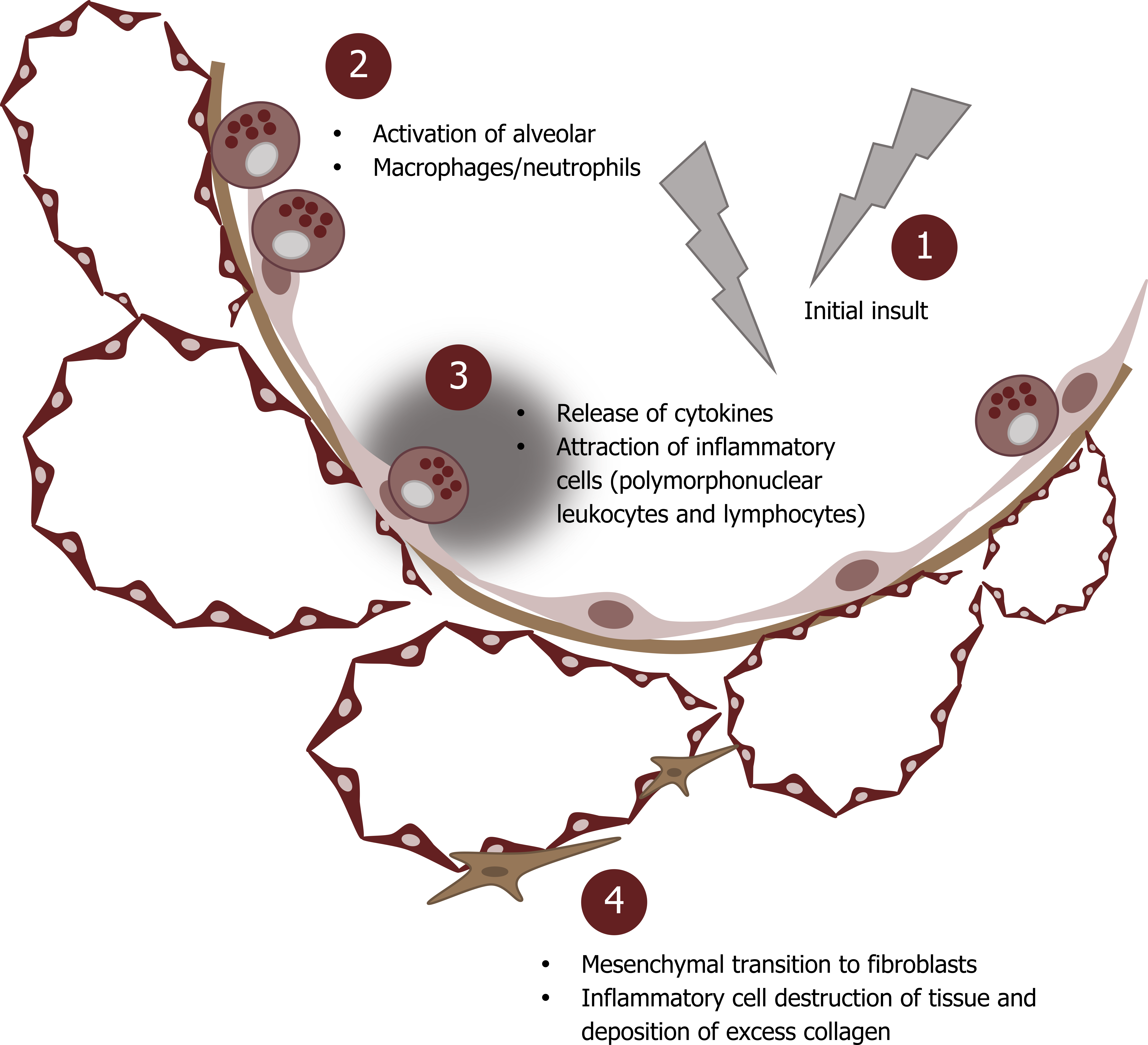
Now we will look at the generalized mechanism of interstitial lung disease (figure 4.1). It is worth noting that the numerous conditions that the term ILD encompasses have subtle differences in mechanism and manifestations, and these differences are what we will deal with elsewhere.
Generally though, ILD starts with an initial insult to the lung (#1, figure 4.1); the type of insult is a major contributor to the different ILD conditions. (It probably will not be what is depicted in figure 4.1, but if it was, it might lead to the inflammatory condition of tiefitis.)
There is then a response by neutrophils and alveolar macrophages (#2, figure 4.1). The macrophage response seems particularly important to the development of ILD. Release of cytokines (#3, figure 4.1) attracts other inflammatory cells, and the arrival of polymorphonuclear leukocytes and lymphocytes play an important role in disease instigation. These cells release cytokines, enzymes, and toxic oxygen radicals that damage and destroy local tissue. Released growth factors, such as TGF-Beta, instigate the transition of mesenchymal cells to fibroblasts.
It is worth noting at this point that some forms of ILD are caused by an exaggerated immune reaction—either through an allergic-like response, or a direct immune disorder.
The destruction of tissue and activity of a growing number of fibroblasts results in the inflamed interstitium becoming fibrosed with excess connective tissue, particularly collagen (#4, figure 4.1).
Pathology of ILD
These changes in structure dramatically change the functional and mechanical properties of the tissue. The changes also tend to follow a characteristic pattern, although, as you might imagine, different conditions have subtle differences in pattern.
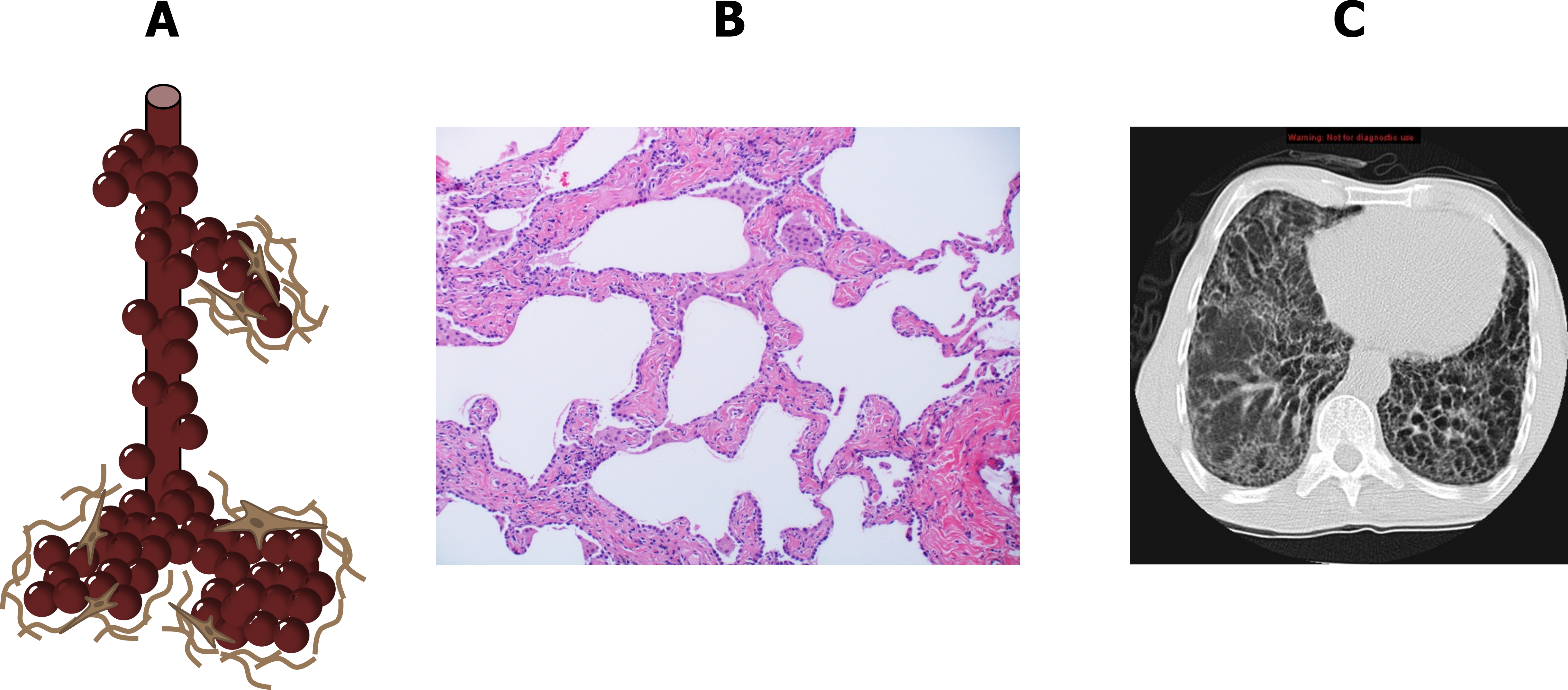
The action of fibroblasts laying down connective tissue, combined with the destruction of alveolar and capillary structures, leads to a widening of airspaces with thick collagenous and infiltrated walls (figure 4.2A and 4.2B), which are a functionally significant departure from the ideal structure for gas exchange.
The thickened basement membrane poses a significant obstacle to the transfer of gases, and the dense connective tissue stiffens the lung and thereby reduces its compliance. Combined with loss of capillary beds and airspace surface area, gas exchange is reduced.
At the end of the disease the lung takes on a characteristic honeycomb appearance, and “ground glass opacities” are a hallmark sign on CT images (figure 4.2C). These morphological changes lead to pathophysiological consequences that are shared by most forms of the disease, as all cause varying degrees of interstitial inflammation and connective tissue deposition.
Pathophysiology of ILD
The first major issue is the reduced diffusion capacity of the involved areas, and all ILD patients demonstrate a reduced transfer factor, or DLCO (figure 4.3).
Because of the heterogenous distribution of the disease, and the involvement of the pulmonary circulation, severe V/Q abnormalities arise throughout the lung. This and the reduced diffusion capacity result in hypoxemia. In the chronic disease state this may lead to cor pulmonale (figure 4.3).
The reduction in lung compliance leads to a reduced lung volume. This is easily detected with spirometry as shown by the inner plot of the flow-volume loop in figure 4.3. Because FEV1 and FVC are both reduced, the ratio frequently remains the same or may even rise; as such FEV1/FVC is a poor indicator of restrictive disease.
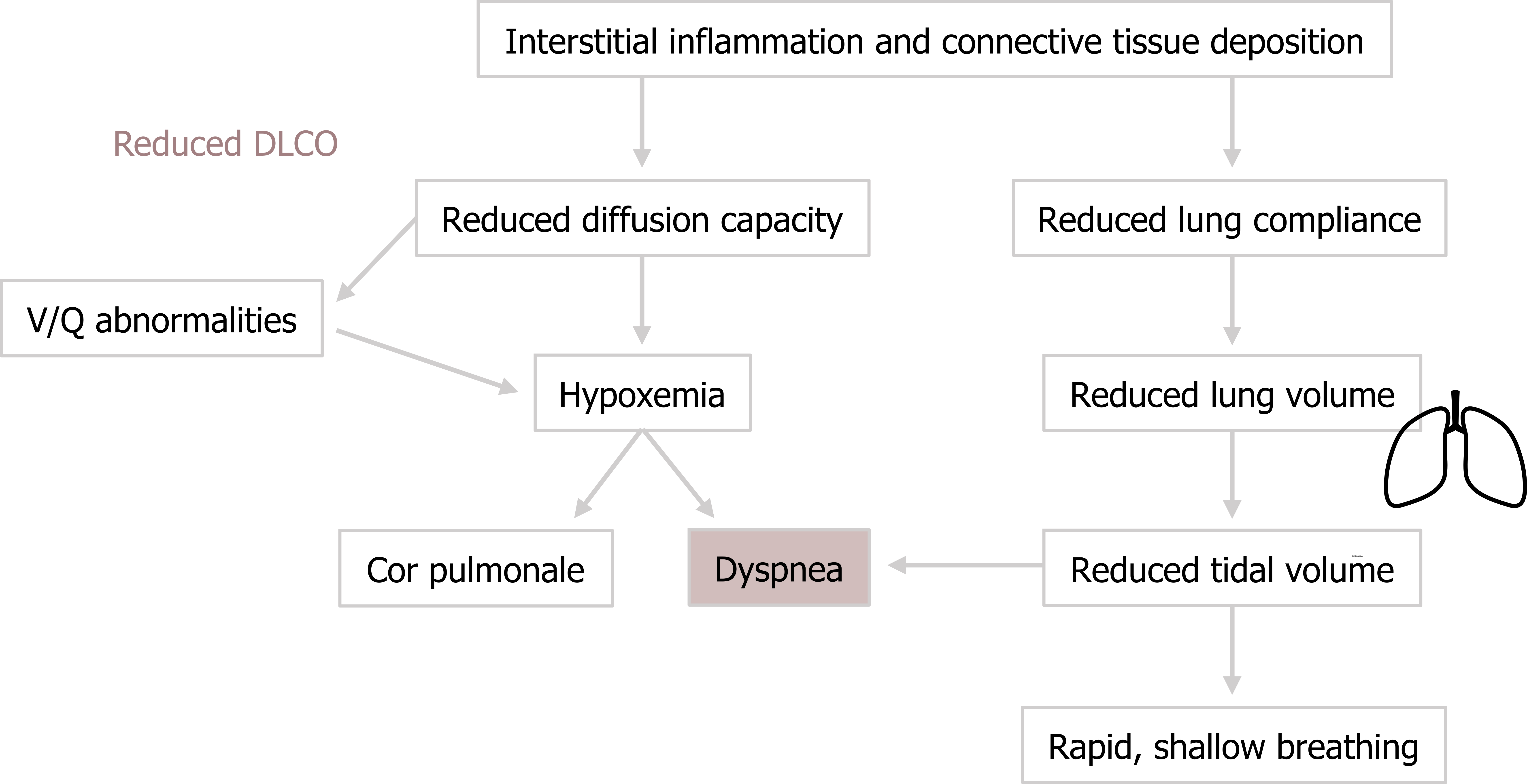
The reduced lung volume and compliance results in a characteristic rapid, shallow breathing pattern as the patient tries to maintain alveolar ventilation (figure 4.3). However, while avoiding unnecessary increases in the work of breathing by not trying to over-expand the noncompliant lung, the rapid shallow breathing proportionally increases dead space ventilation.
The reduction in tidal volume, combined with a raised hypoxic drive to breathe, results in the cardinal symptom of ILD, which is dyspnea (figure 4.3).
Clinical Signs of ILD
The correlation of dyspnea and disease stage is closer in ILD than any other respiratory disease. The onset is insidious, appearing first during exercise, and it likely contributes to the other major complaints of weakness and fatigue. The dyspnea gets progressively worse until it can be debilitating.
Inflammation and excitation of pulmonary receptors leads to a nonproductive and persistent cough, and upon examination patients will have limited chest expansion and demonstrate the characteristic breathing pattern of restrictive lung disease.
Hallmark lung sounds are fine crackles, commonly found at the base of the lung, and may appear louder than expected because of increased transmission through denser than normal tissue.
At later stages of the disease the patient shows signs of the prolonged hypoxemia with digital clubbing and cyanosis.
Forms of Interstitial Lung Disease
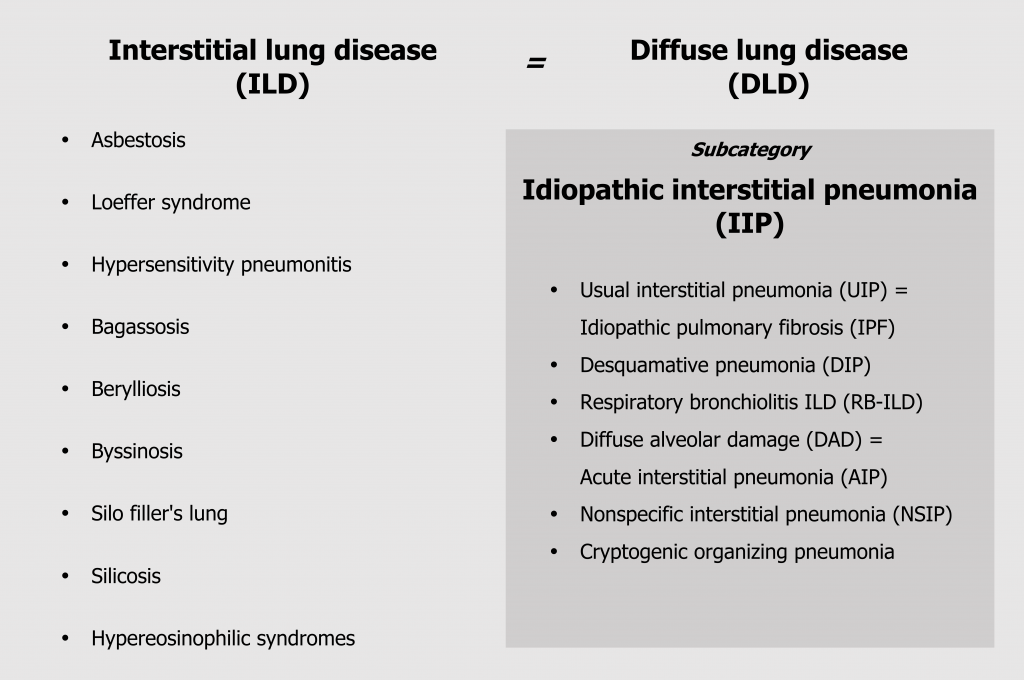
The term interstitial lung disease encompasses about 150 different conditions, and the classification of these conditions is amazingly confusing for numerous reasons. Some classifications are developed by “lumpers” who believe the separate conditions are components of the same spectrum of disorders; other classifications are developed by “splitters,” who believe the conditions are distinct. This is made more confusing by a lack of consistent nomenclature between disciplines for the same condition—and this spanner in our work starts at the highest classification level, where what pathologists refer to as an interstitial lung disease, radiologists call a diffuse lung disease (figure 4.4). Then of course, we will reduce everything to an acronym just to make it even more fun! Convention will likely become to use the pathologist’s nomenclature, but the radiology versions are included here for reference.
Our major subcategory is idiopathic interstitial pneumonia (figure 4.4), or IIP, and this is divided again into six more useful categories that can be distinguished by history, time line, and histological changes. We will deal with these categories in this section, but it might be noted that usual interstitial pneumonia, still frequently called idiopathic pulmonary fibrosis, is the only one that remains untreatable, and early differentiation from the other forms is critical (figure 4.4).
Interstitial lung diseases can also be induced by numerous different environmental causes that produce nuanced conditions that are distinguishable by environmental and social history (figure 4.4) as well as specific histological features.
The characteristics of usual interstitial pneumonia have been covered in the “Basis of ILD” section, so let us start looking at the pathophysiological and clinical features of the other broader disease categories.
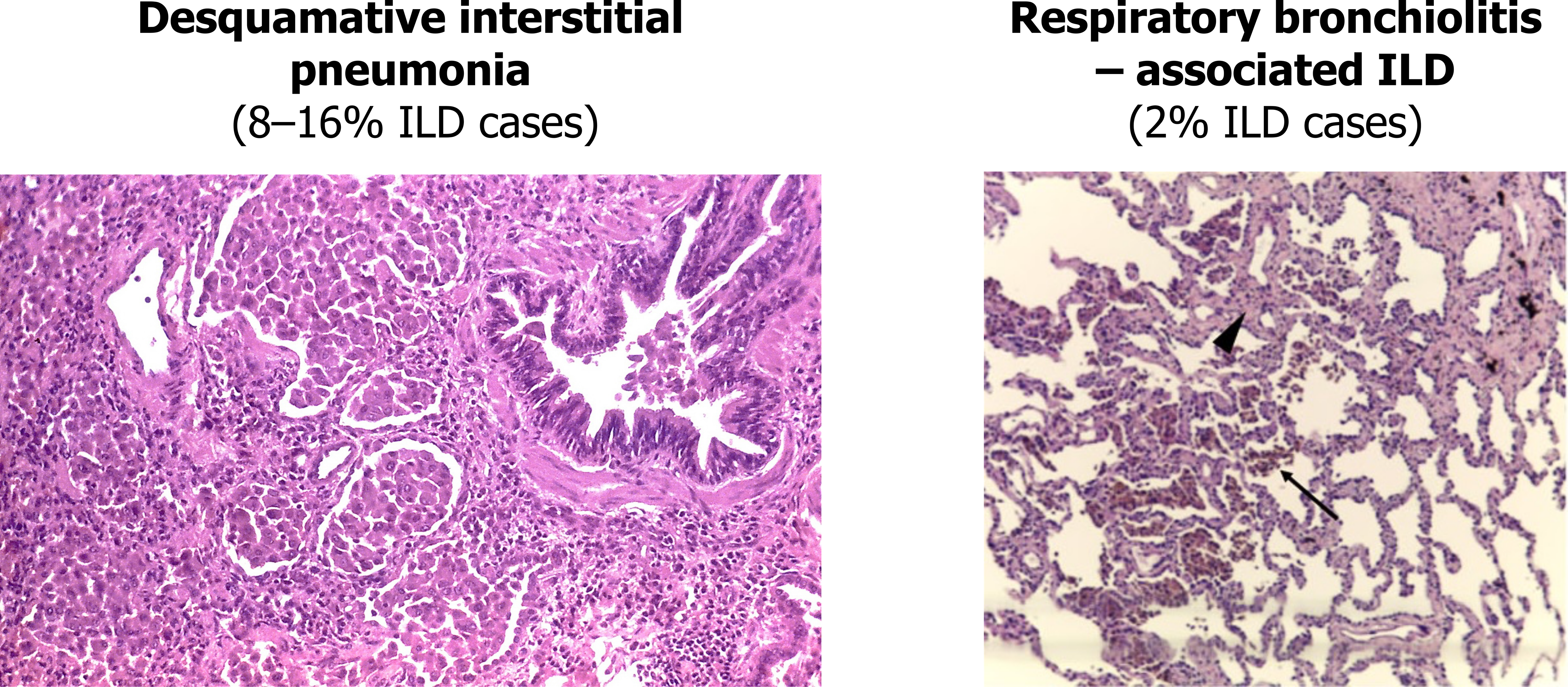
Desquamative Interstitial Pneumonia and Respiratory Bronchiolitis–Associated ILD
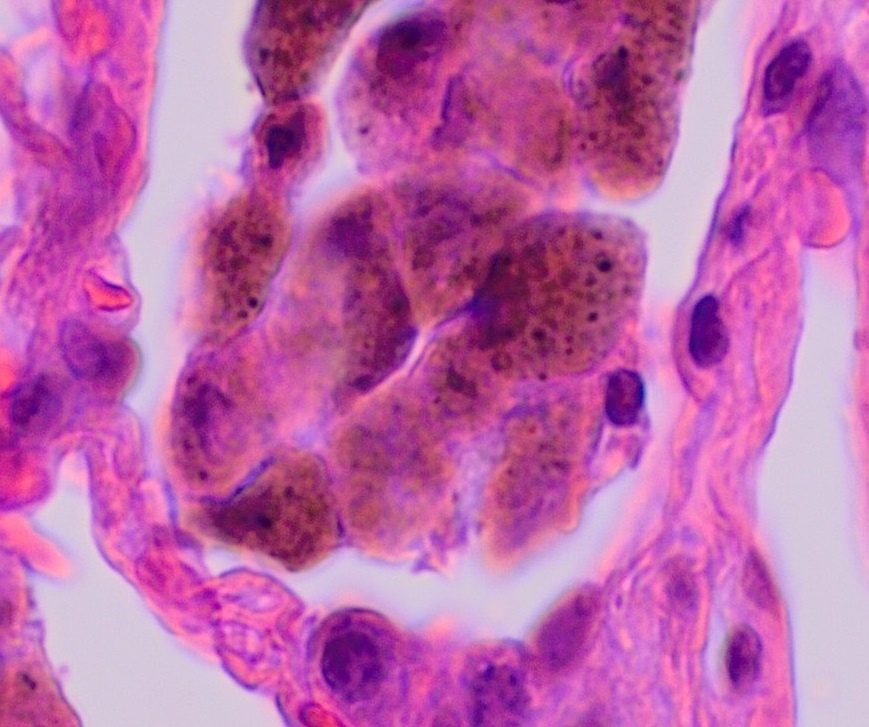
We can deal with the first two together as both share common characteristics and are potentially the same disease occurring in different anatomical locations. Desquamative interstitial pneumonia and respiratory bronchiolitis–associated ILD are both smoking related and are relatively uncommon.
The histological hallmark is accumulation of numerous smoker’s macrophages in the airspaces (figure 4.5) or the first- and second-order respiratory bronchioles. These macrophages have a characteristic brown pigmentation. In desquamative interstitial pneumonia the airspaces are the primary site of involvement, whereas a respiratory bronchiolitis–associated ILD sees more involvement of the bronchioles (as the name suggests). The alveolar septum may be thickened with infiltrate and there may be mild peribronchilor or alveolar fibrosis, but this does not result in a honeycomb pattern seen in usual interstitial pneumonia.
These ILDs are more prevalent in men, and are usually found in the fifth decade of life and after thirty-pack years. They are marked by the gradual and insidious onset of dyspnea, but lung reductions are usually minimal with both forms. The response to corticosteroid therapy and smoking cessation is good in about 80 percent of patients who remain stable or improve.
Diffuse Alveolar Damage (DAD)
While the development of most interstitial lung diseases is slow and insidious, the hallmark of diffuse alveolar damage is rapid, occurring in a matter of days and often in previously healthy individuals. The manifestation of the disease is similar to acute respiratory distress syndrome, and in fact it has been suggested that DAD is a form of ARDS.
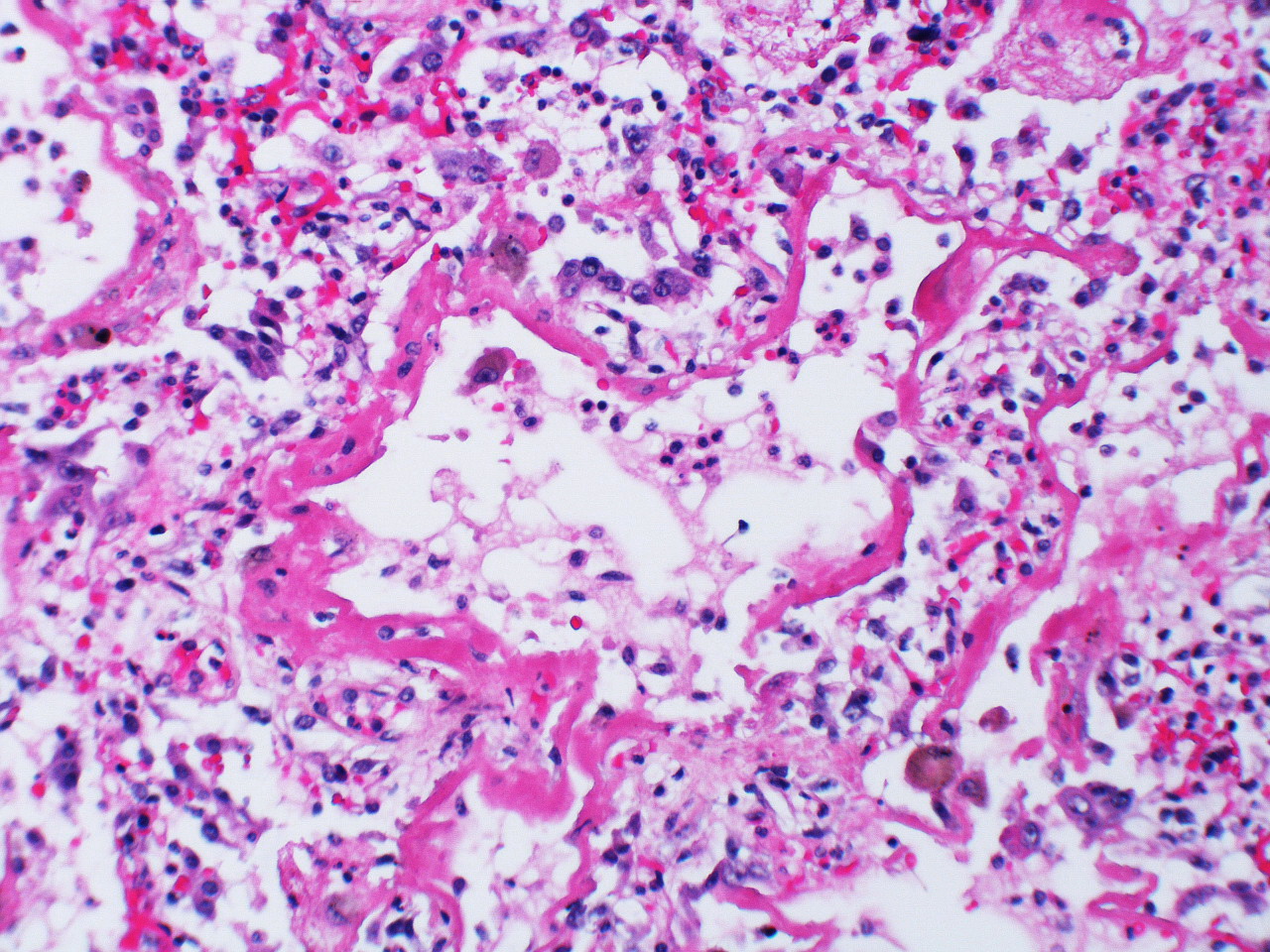
The start is marked by a brief exudative phase with fluid entering the airspaces, but the following organizing or proliferative phase is what is usually seen by the time a biopsy is taken and where the similarities to ARDS are seen. The alveolar septa are thickened due to the interstitial edema and the septa may collapse or appose each other (figure 4.7). There is marked infiltration of the interstitial and airspaces by inflammatory cells, and type II cells proliferate. The destruction of the alveolar structure leaves a sludgy hyaline membrane of debris. Thrombi in small arteries may also be apparent.
Should the patient survive (about 50 percent do not) the healing phase can show recovery of the alveolar structure with varying degrees of fibrosis. Many patients return to normal lung function, but a few show a progressive fibrotic process that resembles idiopathic pulmonary fibrosis.
Without biopsy, DAD is usually differentiated from other forms of interstitial disease by its rapid onset, but this can be confused with acute exacerbations of other diseases. However, the uniform pattern of damage in real DAD is representative of a single time line.
Nonspecific Interstitial Pneumonia
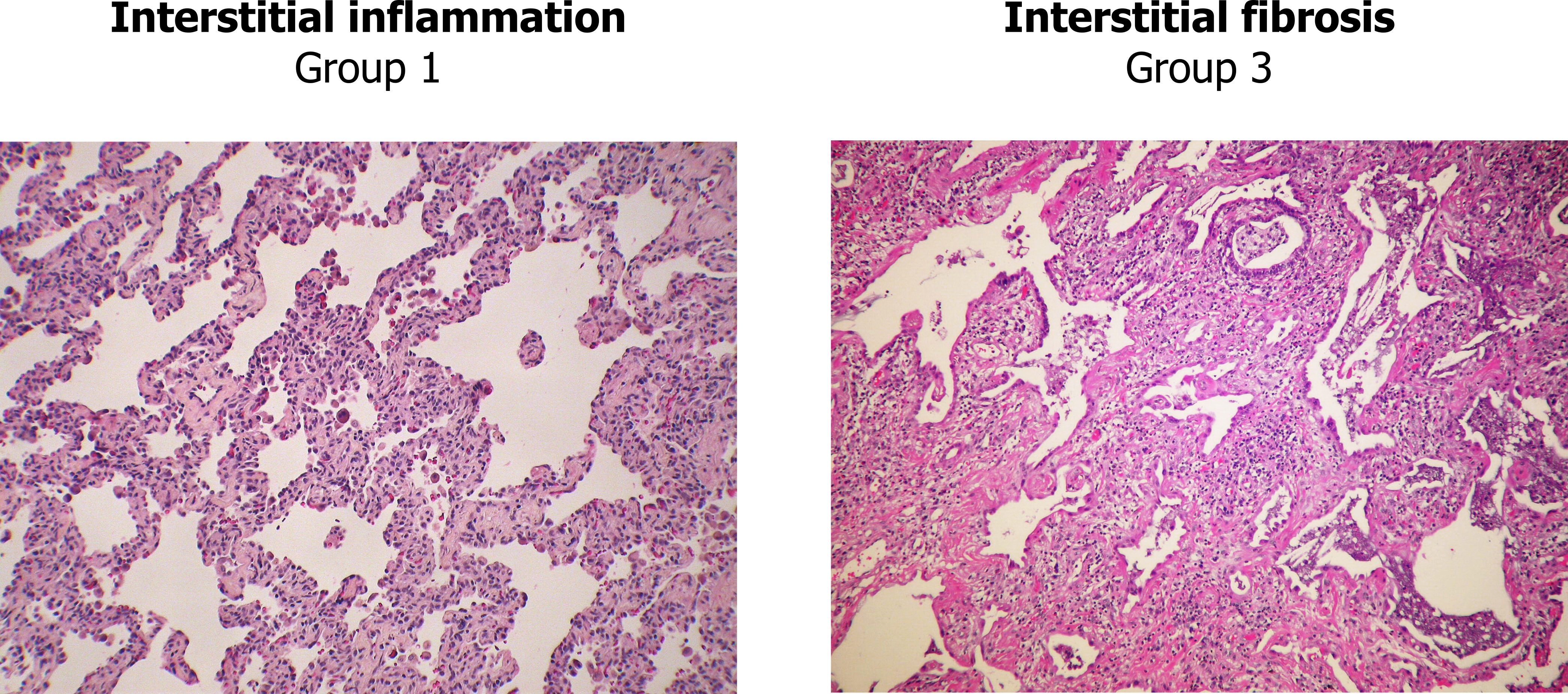
Our next disease is at least courteous enough to only have one name, nonspecific interstitial pneumonia (NSIP), but irritatingly, it has three groups that are determined by the degree of either interstitial inflammation or fibrosis. Group 1 is primarily inflammation, group 2 involves inflammation and fibrosis, and group 3 is primarily fibrosis. The differences in groups are most clearly seen looking at the extremes—group 1 shows the puffy alveolar septa infiltrated with lymphocytes (left panel, figure 4.8), whereas group 3 shows a matrix of fibrosis that can be distinguished from usual interstitial pneumonia by the absence of fibroblastic foci and a homogenous onset and distribution (right panel, figure 4.8). As its name suggests, the distinguishing feature of nonspecific interstitial pneumonia is the lack of features that determine it to be something else. If that sounds a bit wishy-washy, take solace in the fact that even experts argue over its classification.
The presence of lymphocytes in biopsy and bronchoalveolar lavage fluid suggests the involvement of the immune system in the pathogenesis of nonspecific interstitial pneumonia. This is supported by the occurrence of NSIP in immune diseases such as HIV infection and several connective tissue disorders including polymyositis, rheumatoid arthritis, and systemic sclerosis. Our understanding of the pathological mechanisms is still evolving.
Cryptogenic Organizing Pneumonia
Our final major classification is cryptogenic organizing pneumonia (COP). This form of interstitial disease affects the distal bronchioles, respiratory bronchioles, and alveoli, but the primary site of injury is usually the alveolar walls.
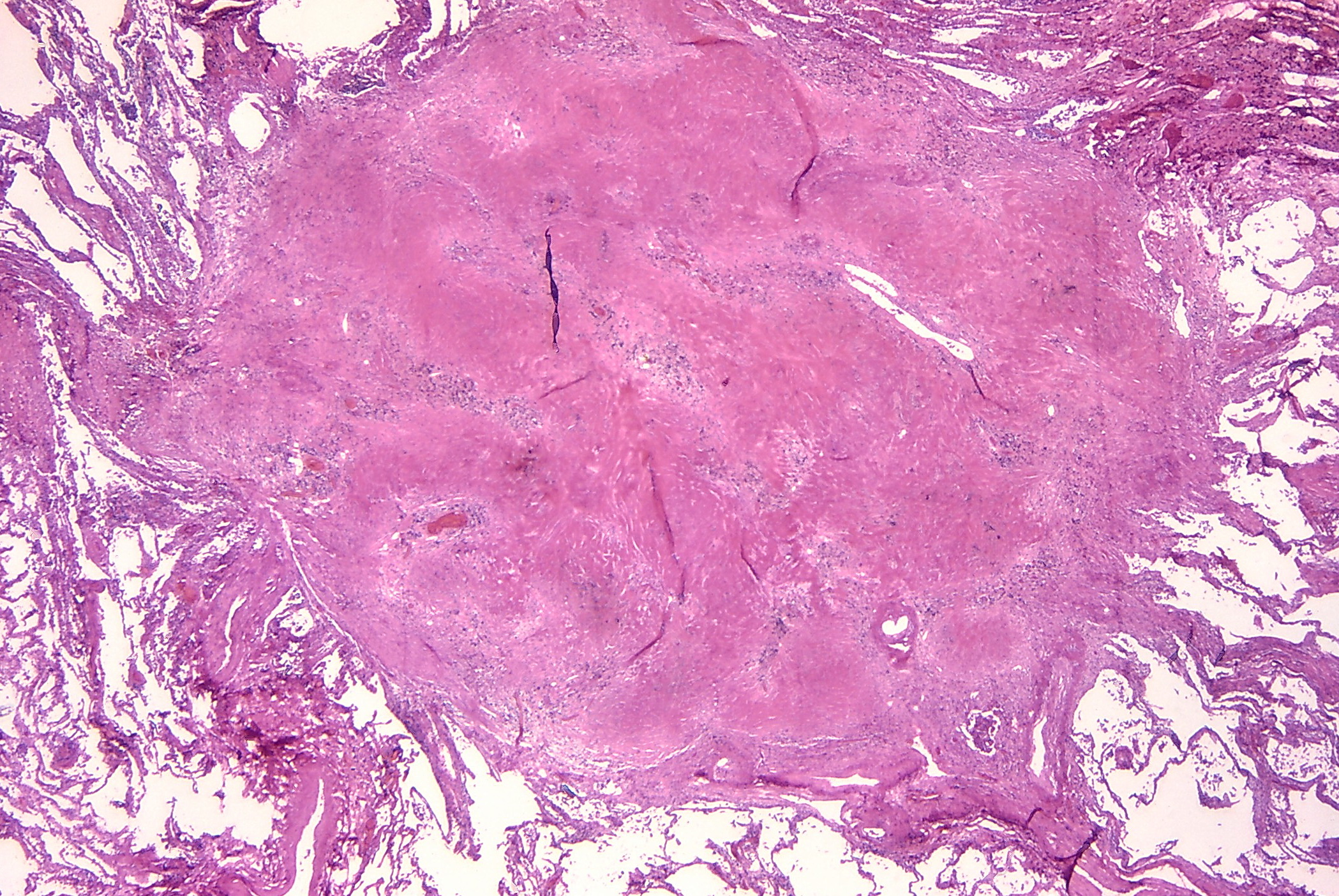
The hallmark of COP is an excessive proliferation of granulation tissue made of collagen-embedded fibroblasts and myofibroblasts that starts in the alveolar space. These plugs of fibrotic tissue may extend from one alveolus to another via the pores of Kohn and give rise to a characteristic butterfly pattern. The pathogenesis is an initial alveolar injury, with plasma proteins leaking into the alveolar lumen that is followed by recruited fibroblasts depositing connective tissue with the lumen itself. These fibrotic lesions show a homogenous time line and movement to the distal airways, but are actually reversible, which is in contrast of the lesions seen in usual interstitial pneumonia. In COP the lung architecture is maintained, probably through more thorough regulation of angiogenesis and apoptosis than that seen in usual interstitial pneumonia (UIP).
The onset of COP is marked with dyspnea and dry cough (as with most ILDs), and it has a moderate time line of a couple of months, after which symptoms subside. History is again important to determine the initial insult, and potential culprits include connective tissue disease, new medications, or exposure to therapeutic radiation, fumes, or dusts.
Environment-Induced ILDs
Now we will look at several specific forms of interstitial disease that are related to occupational exposure. While these forms of ILD have some distinguishing factors, the importance of taking a good history cannot be understated.
Silicosis
Silicosis is related to exposure to silica that occurs frequently in occupations such as stone cutting, foundry work, and mining. Cutting or breaking stone can produce crystalline silica, and when less than 5 microns in diameter, it becomes respirable. When particle size is between 1 and 3 microns, it can reach the alveoli.
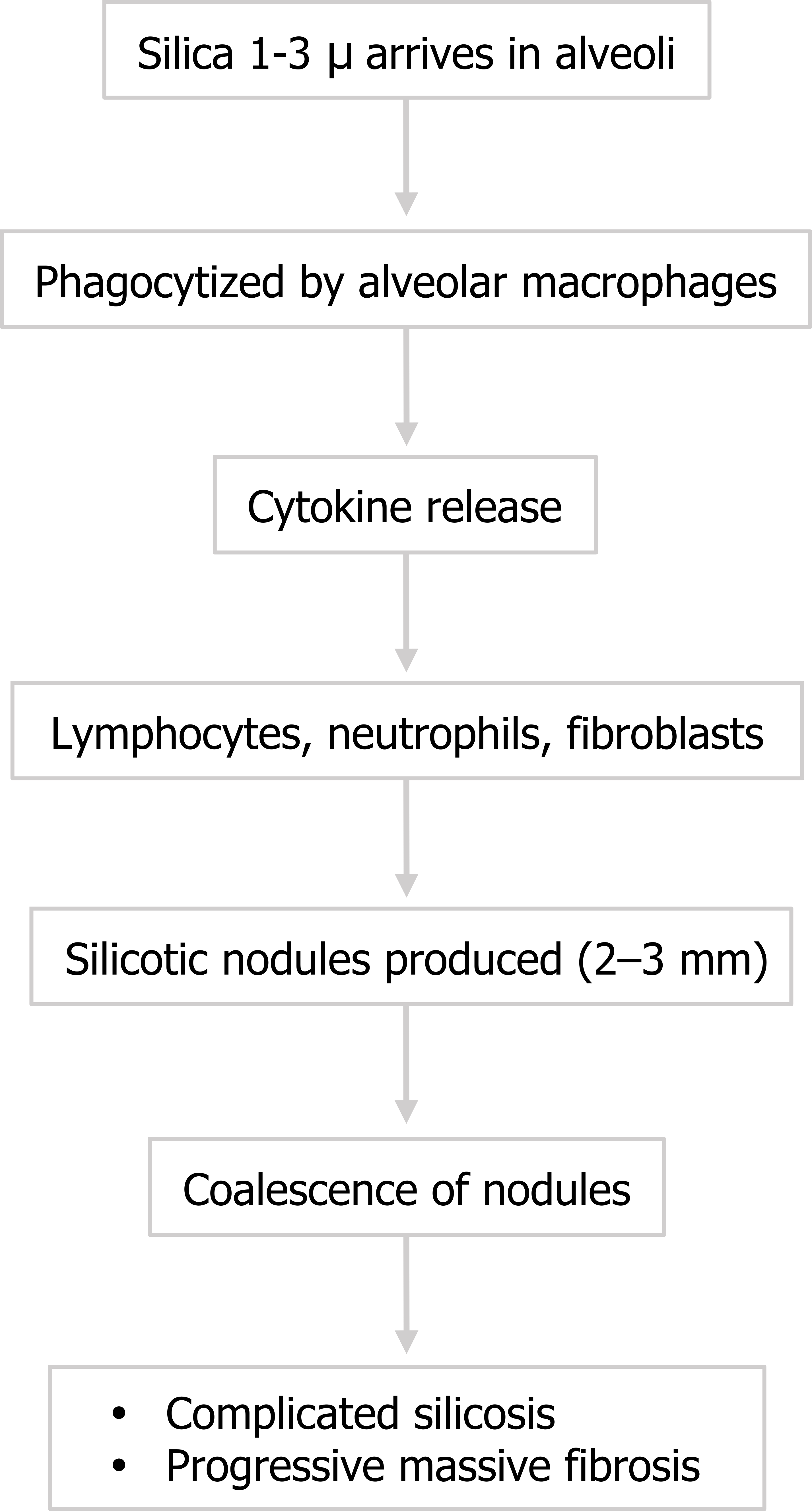
The formation of silicosis can be acute with heavy brief exposure (often seen in sandblasters), or chronic and insidious with more prolonged lighter exposures. The process is initiated with alveolar macrophages engulfing the crystals. In response they release cytokines to attract lymphocytes, neutrophils, and fibroblasts—and a familiar story of tissue destruction and laying down of collagen begins. (You might note at this point that engulfing silica in vitro has been shown to damage macrophages, causing them to release their intracellular enzymes, which may contribute to the destructive mechanism in vivo.)
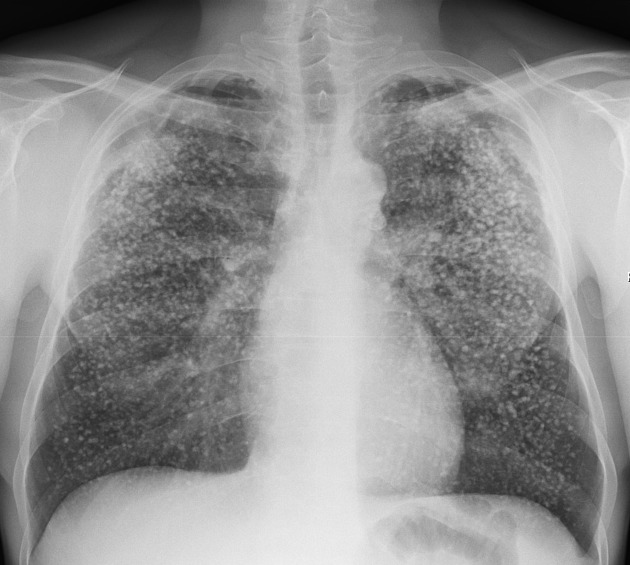
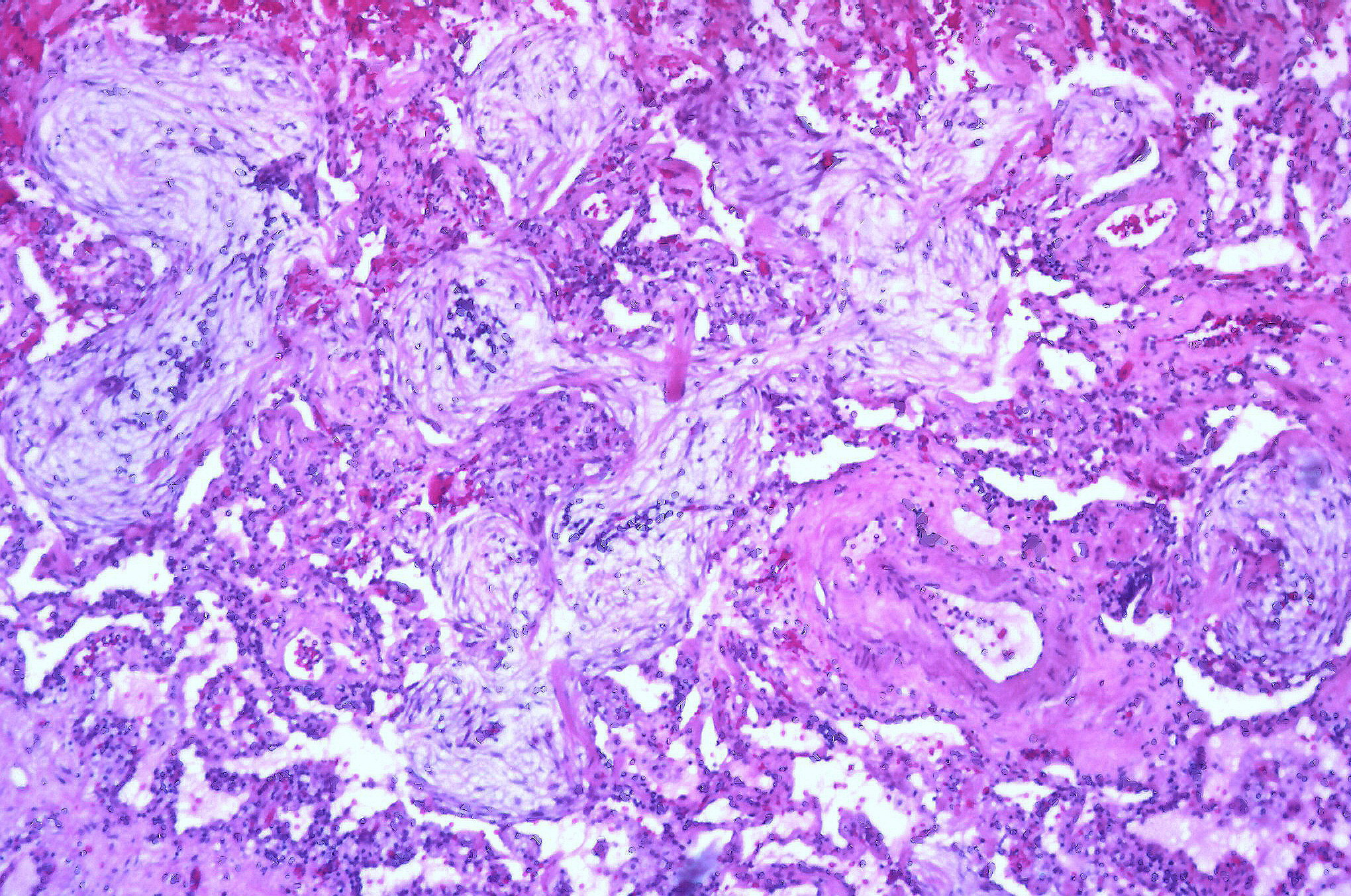
The pattern of collagen deposition is distinct, with silicotic nodules forming with concentric fibers producing a whirled pattern (figure 4.10). These nodules are distributed throughout the lung but are more common in the upper lobes and perihilar area (figure 4.11). They tend to be surrounded by distorted lung tissue that may show emphysematous changes. Ongoing disease coalescence of the nodules produces irregular masses of noncaseating granulomas. This progressive massive fibrosis can be helped with concurrent TB or atypical mycobacterial disease where caseating granulomas may also be present. Likewise silicosis may impair the macrophage response to TB. It causes contraction of the upper lobes and may lead to emphysema in the lower lobes, sometimes with large bullous changes. The pathophysiology of silicosis is summarized in figure 4.12.
After an insidious, asymptomatic beginning, the main symptom of silicosis is dyspnea, with or without cough (cough is likely generated by concurrent smoking). The dyspnea is progressive but other symptoms that occur are often due to secondary, superimposed infection making repeated bacteriological studies important.
The pathophysiology of silicosis is summarized in figure 4.12.
Asbestosis
There are a number of pulmonary manifestations arising from exposure to asbestos. Previously used in the construction and manufacturing industries, the occurrence of related illness led to legislation to restrict its use. However, demolition or renovation of asbestos-containing buildings can still lead to air-born asbestos exposure. The pulmonary manifestations include pulmonary fibrosis, bronchogenic carcinoma, pleural effusion, pleural fibrosis, and mesothelioma. We will deal with the pulmonary fibrosis here and what is known as asbestosis.
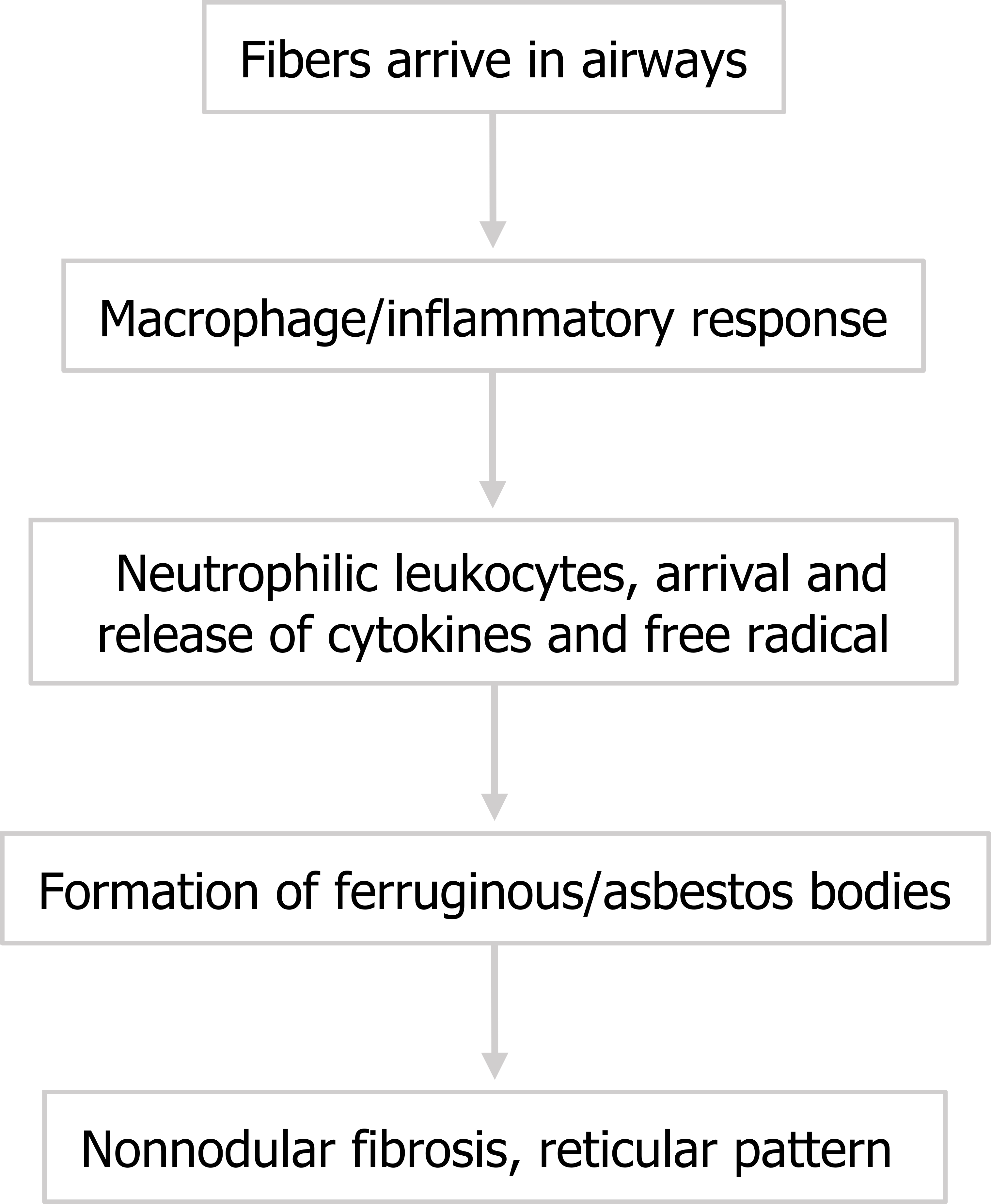
The disease course (summarized in figure 4.13) is similar to that described for silicosis. Asbestos fibers arrive in the alveoli and macrophages initiate an inflammatory response. Note that the arrival of neutrophilic leukocytes and their release of cytokines and oxygen radicals seem to play a significant role. Short fibers can be phagocytized and removed, but larger fibers persist in the airway and perpetuate the inflammatory reaction, promoting fibrosis.
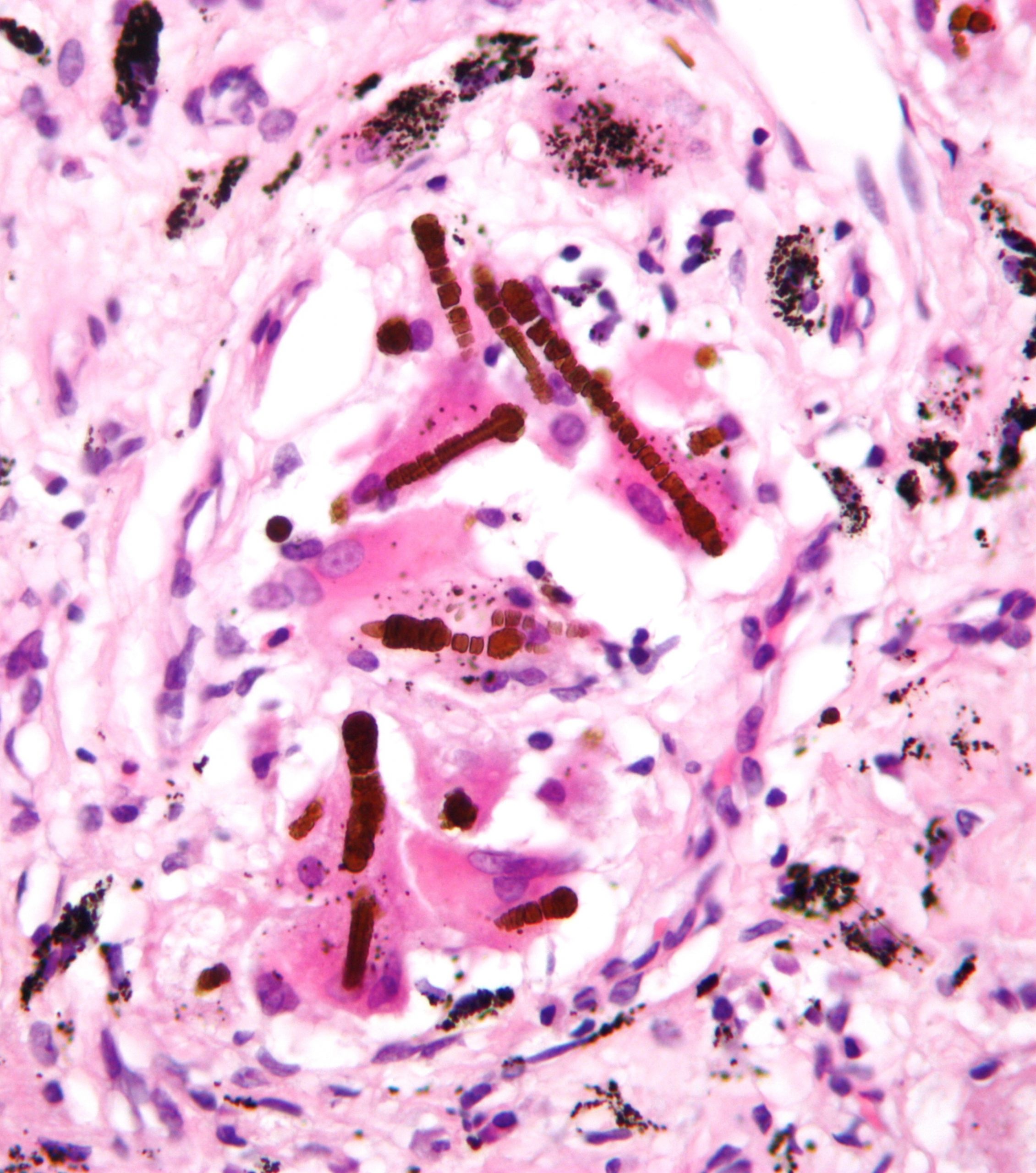
Histologically, these fibers can been seen as asbestos bodies, or ferruginous bodies, as they are coated with iron-containing protein (figure 4.14).
Fibrosis ensues, but in contrast to silicosis, asbestos-related fibrosis is nonnodular and mostly involves the lower lung fields and frequently includes pleural thickening.
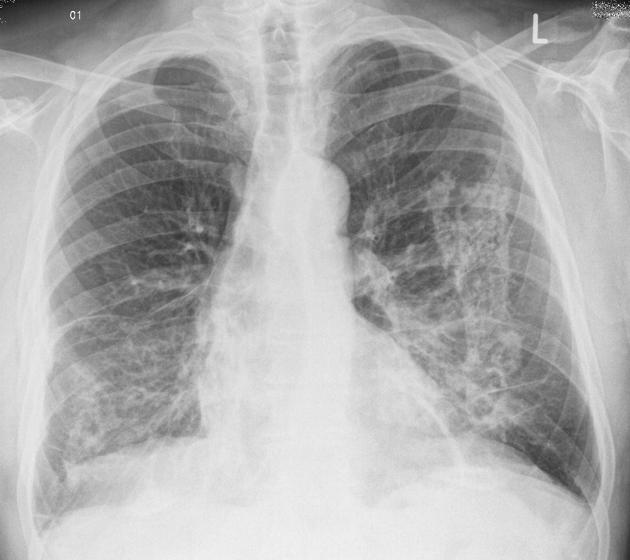
The extent of fibrosis is highly variable, from thickened alveolar septum to complete destruction of the alveolar spaces. In the advanced disease honeycomb lung can be observed with CT. Radiography of later-stage disease shows reticular interstitial markings in the lower lung fields (left panel, figure 4.15). Pleural changes are also more common. Rounded atelectasis may occur after a pleural effusion has been reabsorbed and caused a section of the airway to become trapped. A rounded atelectasis is indicated by the arrow in figure 4.15, and care should be taken not to mistake this for a neoplasm. Abestosis is a risk factor for the development of mesothelioma and should be considered for patients working in “at-risk” environments or occupations.
Coal worker’s pneumoconiosis (CWP)
CWP arises after prolonged exposure to coal dust. While drilling through rock the miner may be susceptible to silicosis, but prolonged and heavy exposure to aerosolized carbon (that is not usually fibrogenic in lesser exposures) can result in its own distinct condition. Even then it can take ten to twelve years of underground exposure to develop.
Again we see the process start with phagocytosis of the coal dust by macrophages after the mucocillary escalator is overwhelmed. The macrophages launch their inflammatory process, and tissue damage is caused by the resultant cytokine bloom and oxygen radical and enzyme release. Fibroblasts form reticulin networks, but there is no significant collagen deposition. Aggregates of reticulin fibers, macrophages, and dust form coal macules (figure 4.16). The coal macules appear as black spots in lung sections and give rise to the condition’s nickname of “black lung.”

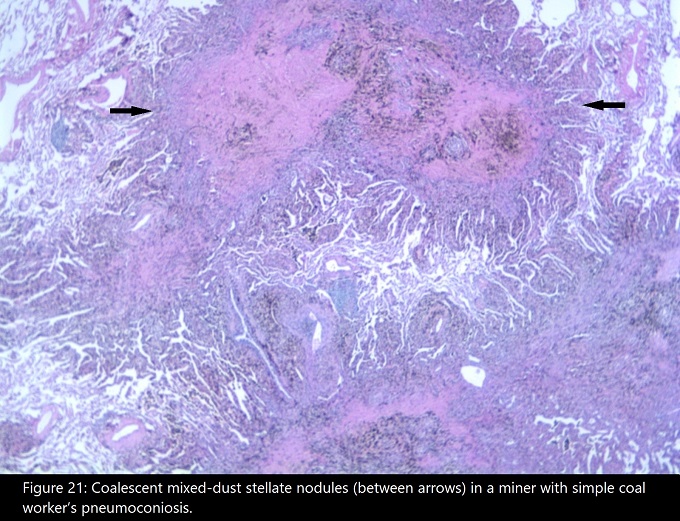
The coal macules are associated with dilation of the respiratory bronchioles that can manifest as focal centrilobar emphysema (figure 4.16). This is referred to as simple CWP, whereas the less common, complicated form involves progressive massive fibrosis, usually in the upper lobes, as in silicosis. However, in CWP these lesions are black and relatively homogenous, where as in silicosis they are a conglomeration of intersecting nodules. Figure 4.17 shows a large black fibrotic lesion destroying the perihilar lung parenchyma.
Clinical manifestations are often complicated by concurrent cigarette smoking that may alone explain the frequency of chronic bronchitis in CWP patients. The simple form can be asymptomatic, but the complicated form produces dyspnea and signs of respiratory failures, pulmonary hypertension, and cor pulmonale.
Berylliosis
The last occupational disorder we will look at is berylliosis, or chronic beryllium disease (CBD), that occurs after exposure to beryllium, a metal used in manufacturing. Here the start to our story is a little different. Beryllium arrives in the airway and there is a hypersensitization of T cells. On subsequent exposures the T cells proliferate—the bronchoalveolar lavage (BAL) fluid of berylliosis patients is rich in sensitized CD4+ cells.
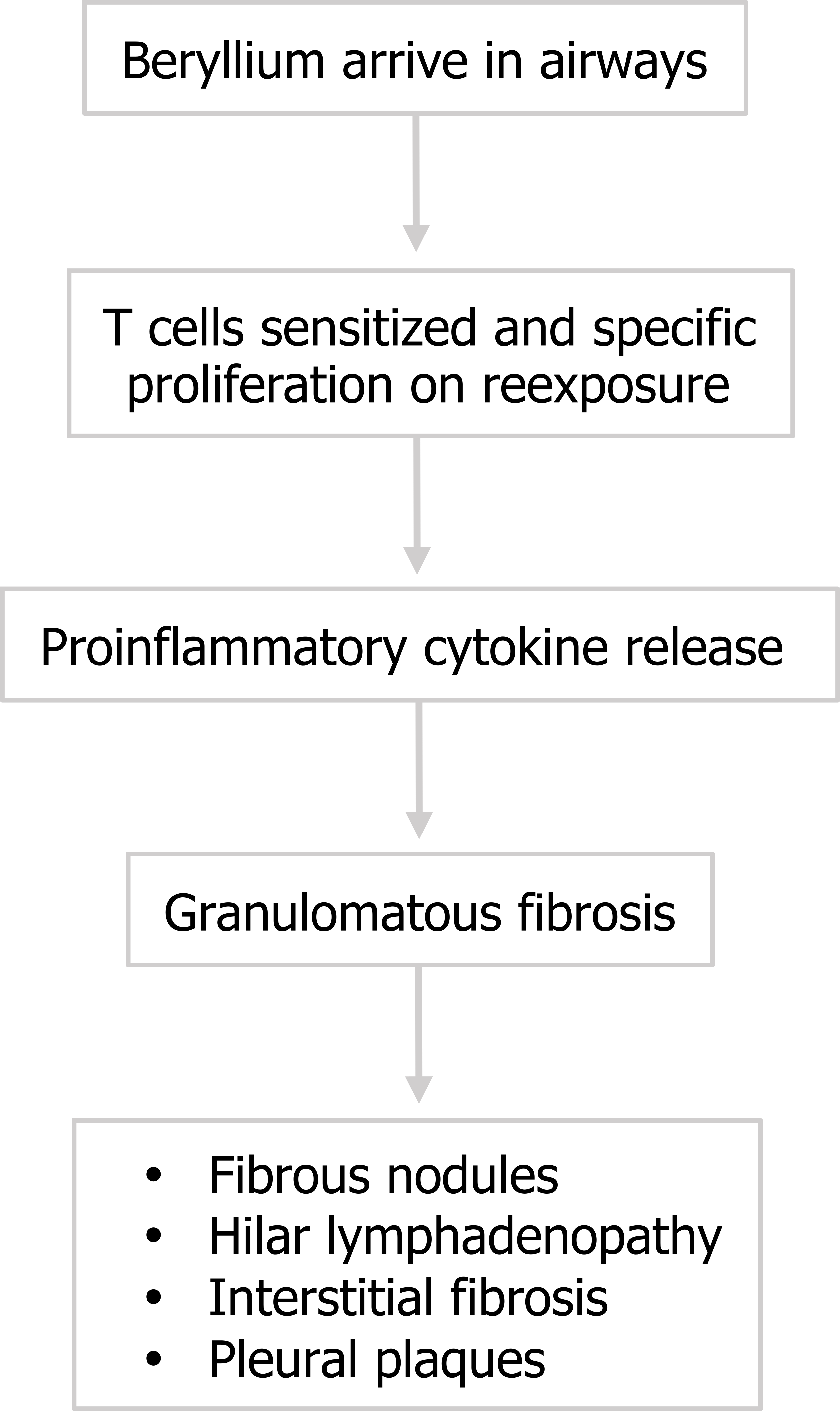
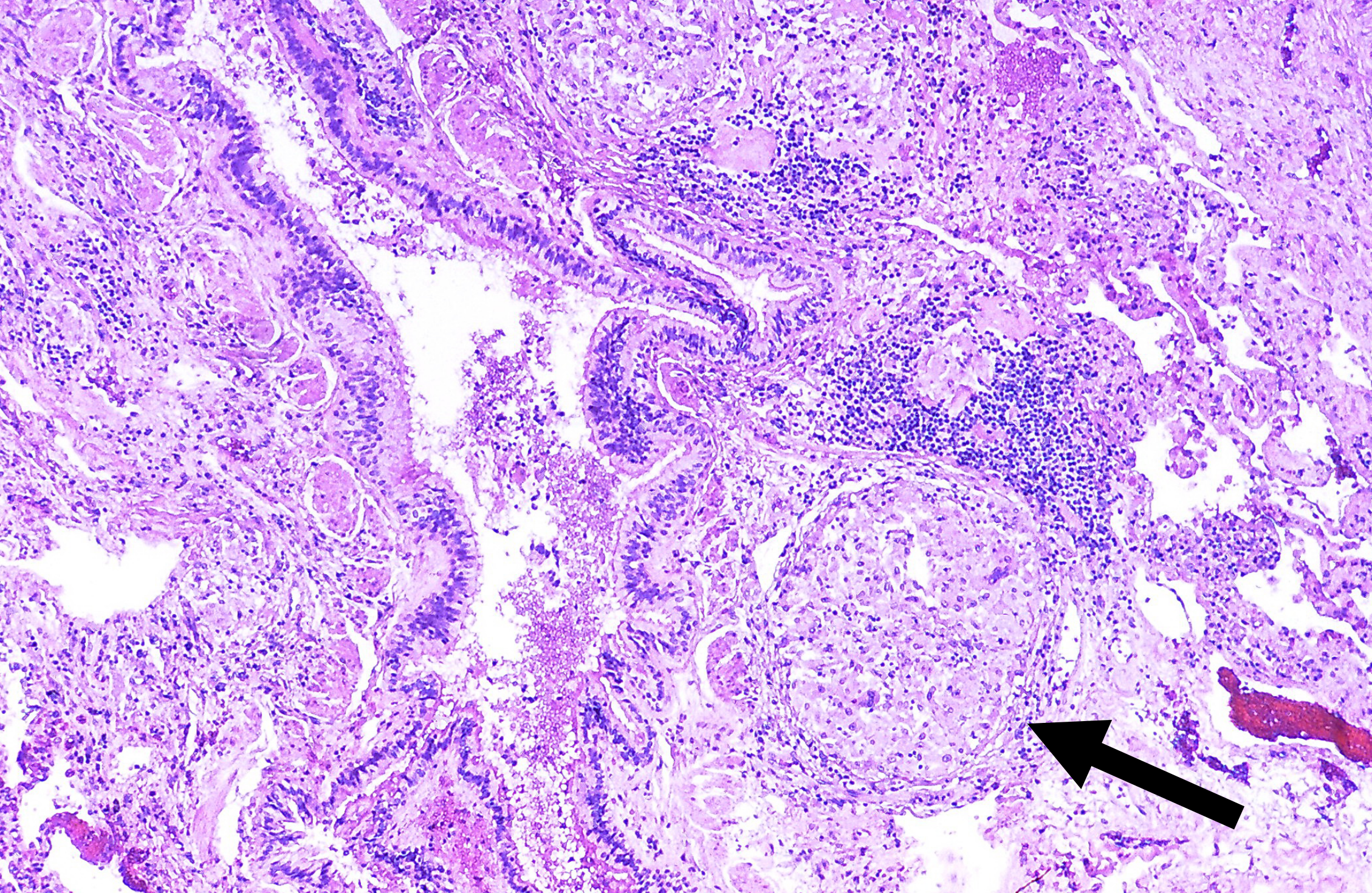
Now we return to our pattern: the abundant CD4+ cells release proinflammatory cytokines and granulomatous fibrosis occurs (figure 4.18). The granulomas (figure 4.19) are indistinguishable from those caused by sarcoidosis (which are also caused by CD4+ cells), and many CBD patients may be misdiagnosed as sarcoidosis cases, so appropriate history taking is paramount. Usually CBD involves greater interstitial inflammation, but the most definitive diagnosis comes from the beryllium lymphocyte proliferation test. The test involves exposing lymphocytes from the patient’s blood or BAL fluid to different concentrations of beryllium and assaying their proliferation.
Susceptibility to becoming hypersensitized appears to have a significant genetic component. Why the process continues after exposure has stopped is unclear, but possibilities include a fundamental T cell disorder, or the fact that the insoluble beryllium causes apoptosis of macrophages, leading them to release a previously phagocytized beryllium load.
As the disease progresses, radiographic findings show that the granuoles can become more organized to produce fibrous nodules that may begin to impact lung function. The immune system involvement can produce hilar lymphadenopathy, and common later signs include interstital fibrosis and pleural thickening.
Summary
So there is a selection of interstitial lung diseases that, while sharing the pathophysiological manifestations of restrictive lung disease, can be distinguished through good history taking or identifying distinct histological features.
References, Resources, and Further Reading
Text
Farzan, Sattar, with Doris L. Hunsinger and Mary L. Phillips. “Chapters 12–15.” In A Concise Handbook of Respiratory Diseases. Reston, VA: Reston Publishing Company, 1978.
Husain, Aliya N. “Chapter 15: The Lung.” In Robbins and Cotran Pathologic Basis of Disease, 9th ed., edited by Vinay Kumar, Abul K. Abbas, and John C. Aster. Philadelphia: Saunders, an imprint of Elsevier Inc., 2015.
Figures
Figure 4.1: Basic mechanism of interstitial lung disease. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/4.1_20220203
Figure 4.2: Changes in Pulmonary histology (A, B) and gross anatomy (C) with interstitial lung disease. Grey, Kindred. 2022. CC BY-NC-SA 3.0. Added Sphere by Ates Evren Aydinel from Noun Project (CC BY 3.0), Smoking-related interstitial fibrosis – Case 268 by Yale Rosen from Flickr (CC BY-SA 2.0), and Case 1 by Jones, J., Weerakkody, Y., et al. from https://doi.org/10.53347/rID-14479 (CC BY-NC-SA 3.0). https://archive.org/details/4.2_20220203
Figure 4.3: Pathophysiological consequences of ILD. Grey, Kindred. 2022. CC BY 4.0. Added Lungs by regara from Noun Project (CC BY 3.0). https://archive.org/details/4.3_20220203
Figure 4.4: Classifications of ILD. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/4.4_20220203
Figure 4.5: ‘Smoker’s macrophages occupying an airspace. Häggström, Mikael. 2021. Public domain. From WikimediaCommons.
Figure 4.6: Examples of DIP and RB-ILD showing alveolar airspace and bronchiolar involvement in each condition respectively. Grey, Kindred. 2022. CC BY-SA 2.0. Added Desquamative interstitial pneumonia by Yale Rosen from Flickr (CC BY-SA 2.0) and Histopathology of respiratory bronchiolitis by Sousa, C., Rodrigues, M., Carvalho, A. et al. from WikimediaCommons (CC BY 4.0). https://archive.org/details/4.6_20220203
Figure 4.7: Diffuse alveolar damage. Rosen, Yale. 2009. CC BY-SA 2.0. From WikimediaCommons.
Figure 4.8: Extremes of non-specific interstitial pneumonia. Grey, Kindred. 2022. Added Non-specfic interstitial pneumonia (NSIP), cellular variant Case 137 by Yale Rosen from Flickr (CC BY-SA 2.0) and Non-specfic interstitial pneumonia (NSIP), fibrosing variant Case 138 by Yale Rosen from Flickr (CC BY-SA 2.0). https://archive.org/details/4.8_20220203
Figure 4.9: Distal airways effected by COP showing butterfly-shaped fibrotic lesions. Rosen, Yale. 2010. CC BY-SA 2.0. From Flickr.
Figure 4.10: Silicotic nodule in parenchyma of lung. Rosen, Yale. 2012. CC BY-SA 2.0. From WikimediaCommons.
Figure 4.11: Silicotic nodules in right upper lobe indicated by arrows. Radswiki, T., Weerakkody, Y., et al. 2020. CC BY-NC-SA 3.0. Case 12 from https://doi.org/10.53347/rID-12513.
Figure 4.12: The pathophysiology of silicosis. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/4.12_20220203
Figure 4.13: Pathophysiology of asbestosis. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/4.13_20220203
Figure 4.14: Ferrugenous bodies associated with asbestosis. Nephron. 2009. CC BY-SA 3.0. From WikimediaCommons.
Figure 4.15: Radiographic finding in asbestosis. Weerakkody, Y., et al. 2021. CC BY-NC-SA 3.0. “Case 4” from https://doi.org/10.53347/rID-7465.
Figure 4.16: Example of Coal Macules in simple CWP. CDC. 2020. Public domain. Figure 21 from CDC.
Figure 4.17: Large peri-hilar lesion in complicated CWP. CDC. 2020. Public domain. Figure 27 from CDC.
Figure 4.18: Pathophysiology of berylliosis. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/4.19_20220203
Figure 4.19: Granulomas of berylliosis. Rosen, Yale. 2012. CC BY-SA 2.0. From WikimediaCommons.
