5 Acute Respiratory Distress Syndrome
Learning objectives
- Describe the pathogenesis and pathophysiology of acute respiratory distress syndrome (ARDS).
Before consensus on its name was reached in 1992, acute respiratory distress syndrome, or ARDS, was known by numerous names, such as “shock lung,” “wet lung syndrome,” “white lung syndrome,” and “congestive atelectasis.” These examples of previous names tell you something about ARDS:
- that it has multiple causes, and
- when we see the words “shock,” “wet,” “congestive,” and “white” (in reference to its radiological appearance, figure 5.8), there are likely inflammatory and edema components.
So let us look at the pathophysiological processes underlying what is now universally known as ARDS.
Pathology of ARDS
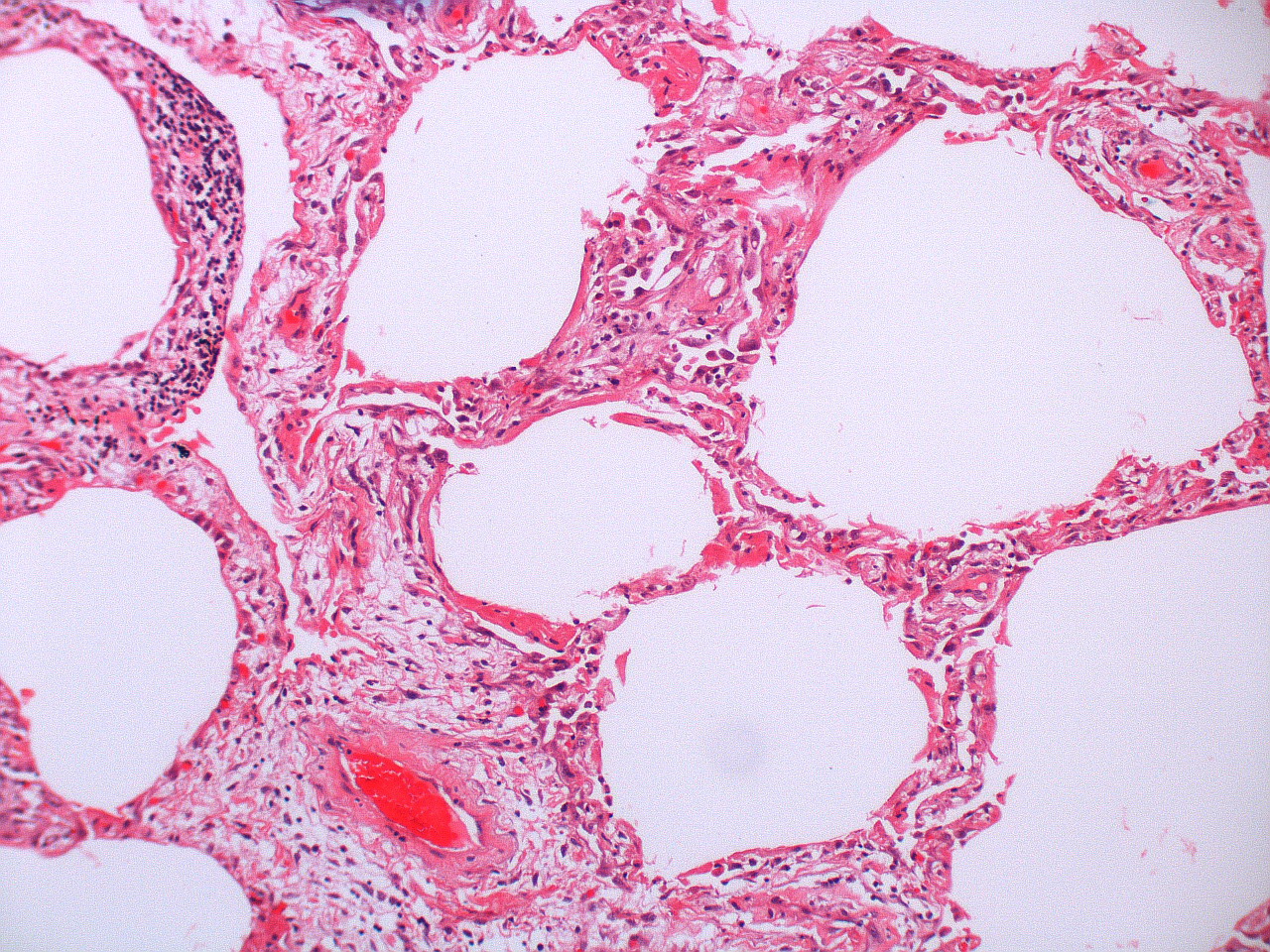
There are about two hundred thousand cases of ARDS in the United States each year. Each case starts with an initial insult to the lung parenchyma and there are numerous examples of this instigating event, but the most common of these (and therefore worth remembering) are sepsis, pulmonary aspiration, and thoracic trauma. The insult can arrive from the airway, such as in pulmonary aspiration or smoke inhalation, or can arrive from the bloodstream, as in a fat embolism or blood-born pathogen.
Regardless of the insult’s route or indeed form, the ensuing pathological events are similar and lead to the same alteration of the lungs. What is initiated is a defensive inflammatory response, and what results is vascular endothelial and alveolar epithelial damage and a leaky alveolar capillary membrane.
Let us look at the process step-by-step.
-
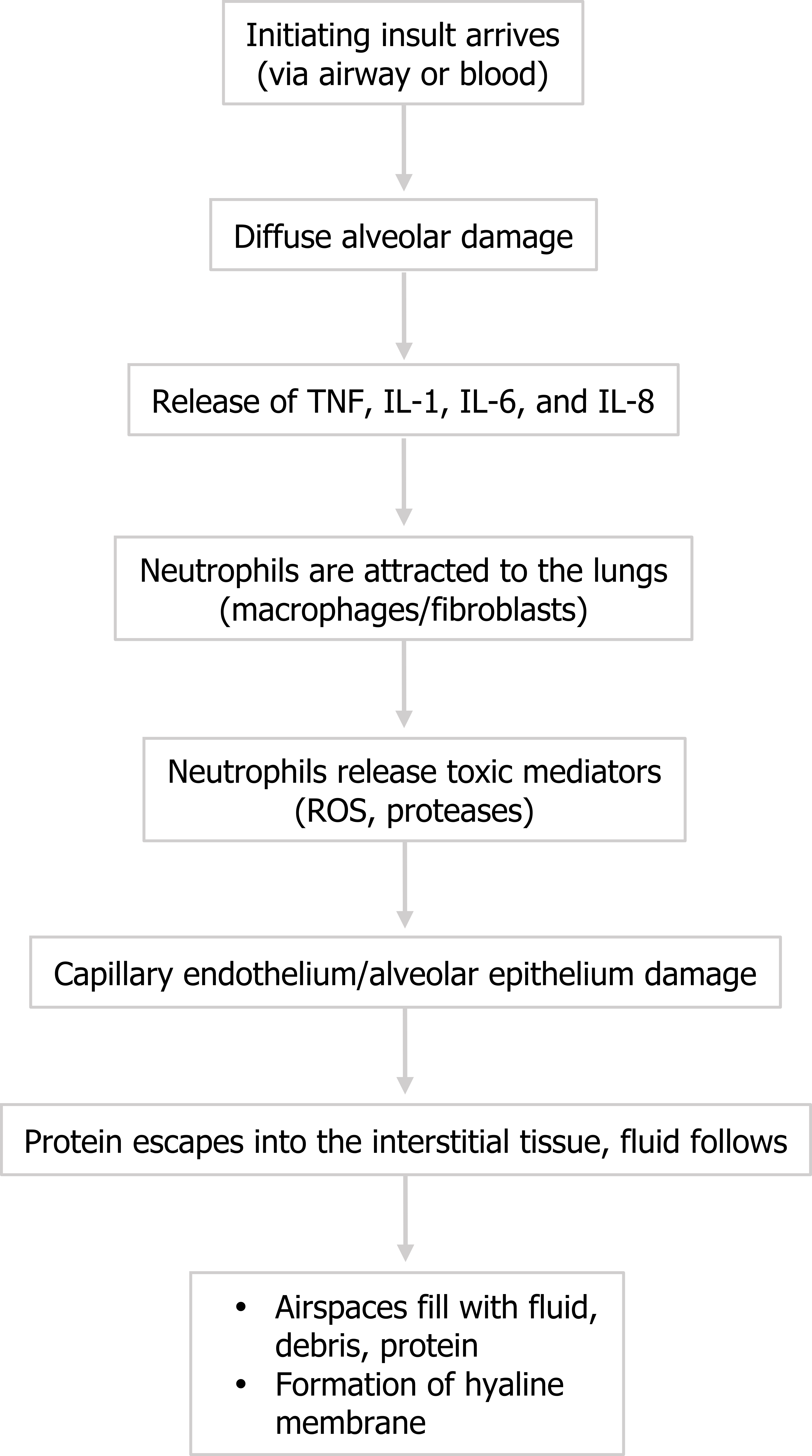
Figure 5.2: Pathophysiological events of ARDS. ROS = reactive oxygen species. The insult (whatever the form) causes diffuse alveolar damage that includes some alveolar septal thickening, hyperplasia of pneumocytes, and formation of eosinophilic hyaline membranes. Despite its name, this “membrane” is not an organized s, purposeful structure, but should be thought of as accumulating debris that is going to block gas exchange.
- The injury causes release of inflammatory cytokines, and tumor necrosis factor, interleukin (IL)-1, IL-6, and IL-8 have been shown to be particularly involved in ARDS.
- The cytokines attract neutrophils to the area, and polymorphonuclear neutrophils play a central role in the inflammatory process. Other cell types can also arrive, such as macrophages, lymphocytes, and fibroblasts, and there is also activation of coagulation and the complement system. Some of the small vessels can be completely obliterated by fibrin-platelet aggregation (figure 5.1), and early proliferation of fibroblasts starts the process of fibrous tissue formation.
- The neutrophils, however, begin to release toxic mediators, but rather than resolve the underlying, initial problem, the released reactive oxygen species and proteases disrupt the capillary endothelium and the alveolar epithelium.
- With these barriers compromised, protein escapes into the interstitial tissue and water follows, and then the compromised alveolar wall can be breached and water enters the airspace.
- Along with water cellular debris and proteins that accumulate in the airspace, providing an oncotic force to draw more water into the airspace, this cellular junk can settles and adds to the hyaline membrane to coat the inner surface of the alveolus, forming a barrier to gas exchange that will persist even after the edema has been resolved.
The process is summarized in figure 5.2.
Stages of ARDS
There are three defined stages for the progression of ARDS.
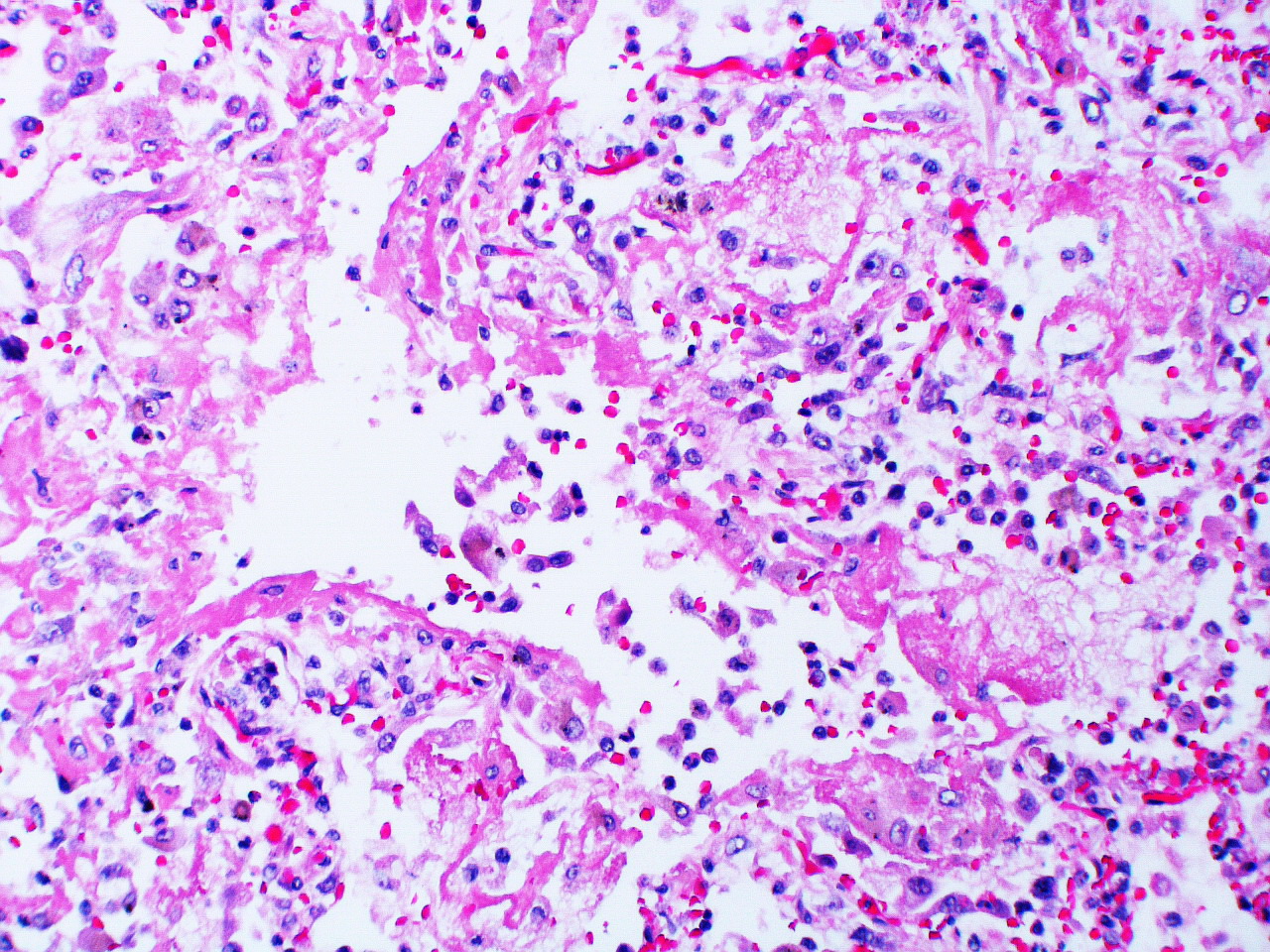
1. Exudative: The first six to seven days comprise the exudative phase that begins with edema appearing in the interstitial walls seen here by the widened alveolar septum. Cellular debris can also be seen in the airspaces, and this can continue with the formation of the hyaline membranes that coat the alveolar surface (figure 5.3).
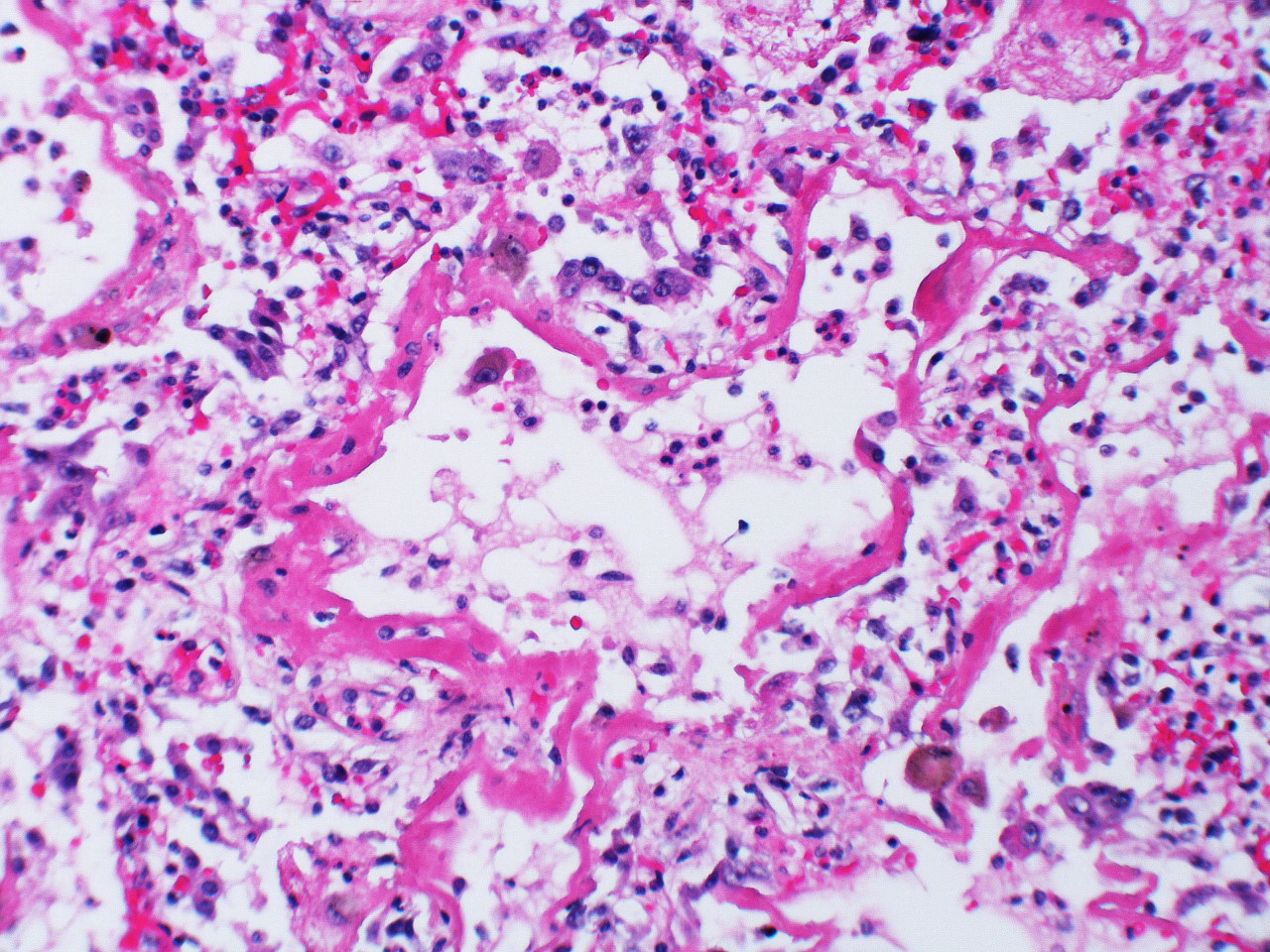
2. Proliferative: In the early proliferative stage, there is increased cell infiltration and squamous metaplasia with proliferation of type II cells (figure 5.4), and these mitotic type II cells have a “hob-nail” like appearance over a time line of weeks. The infiltrating cells include fibroblasts that begin laying down collagen.
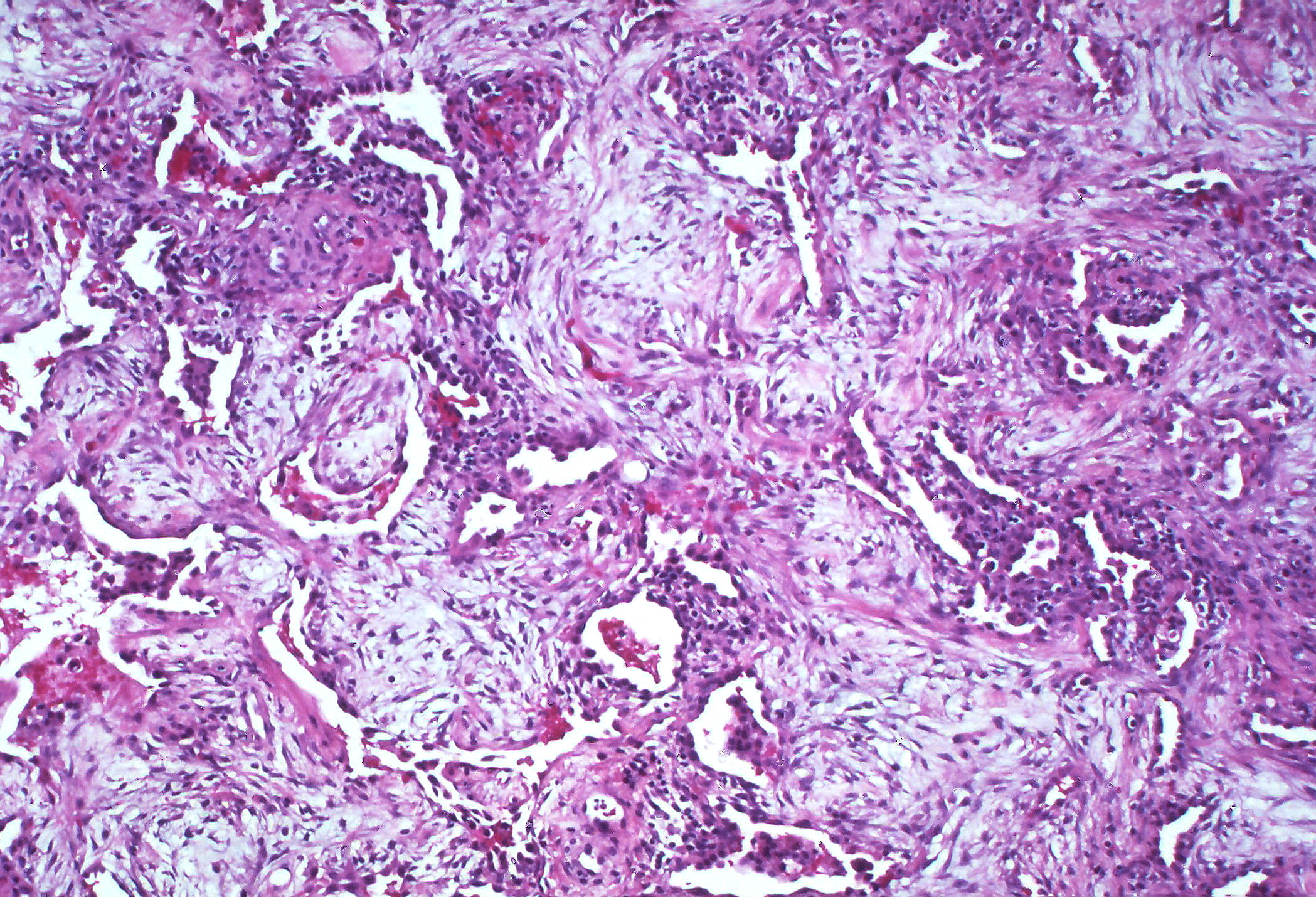
3. Fibrotic: Without resolution the patient may enter the third stage, fibrotic stage, that occurs over months (figure 5.5). This stage occurs much later and is a consequence of unresolved chronic inflammation. Diffuse fibrosis permanently obliterates normal lung architecture and may form cysts.
Systemic pathophysiology of ARDS
We start with initial lung injury leading to alveolarcapillary leaking. As we have seen, this leads to the airspace edema and hyaline membrane formation.
The lack of gas exchange from affected areas produces a right–left shunt and hypoxemia will result. This produces dyspnea, the major symptom of ARDS.
But the disruption caused by the inflammatory process also leads to other pathophysiological issues. The loss of type II cells causes surfactant production to decline. This of course reduces lung compliance, and the resultant increase in the work of breathing contributes to the patient’s dyspnea.
But we are still not done. The fibrin clots forming obstructions in the lung microvasculature lead to V/Q mismatches, and these contribute to the hypoxemia. The obstructed vasculature also produces pulmonary hypertension, which is exacerbated by the vasculature’s response to the hypoxia.
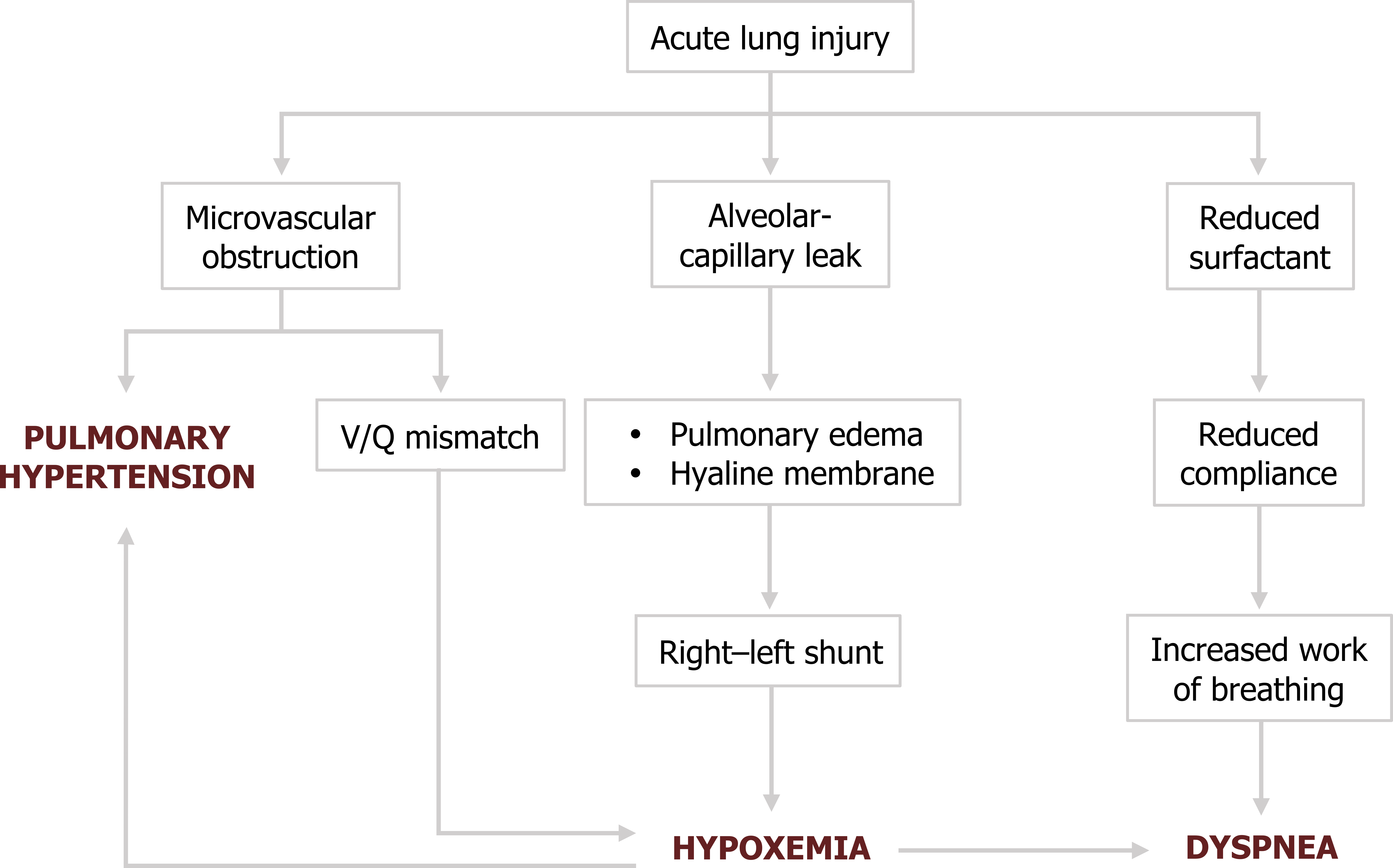
These mechanisms are reflected in the clinical manifestations of ARDS.
Clinical signs of ARDS
The onset of dyspnea usually occurs within one to two days after the initial injury, and as tachypnea arises, this symptom progressively worsens. Cough is common and may produce blood-tinged sputum. Findings on chest exam may be surprisingly scant, but some bronchial breath sounds and crackles may be heard.
As cyanosis becomes apparent, minute ventilation and dyspnea continue to increase and the patient will likely become distressed.
A high ventilatory rate driven by the hypoxemia can produce hypocapnia and a respiratory alkalosis. The arterial pH can be complicated by the underlying disorder, and it is not uncommon for a mixed acid–base disorder to occur with concurrent respiratory alkalosis and metabolic acidosis. At the onset of respiratory failure arterial CO2 will rise and produce a respiratory acidosis.
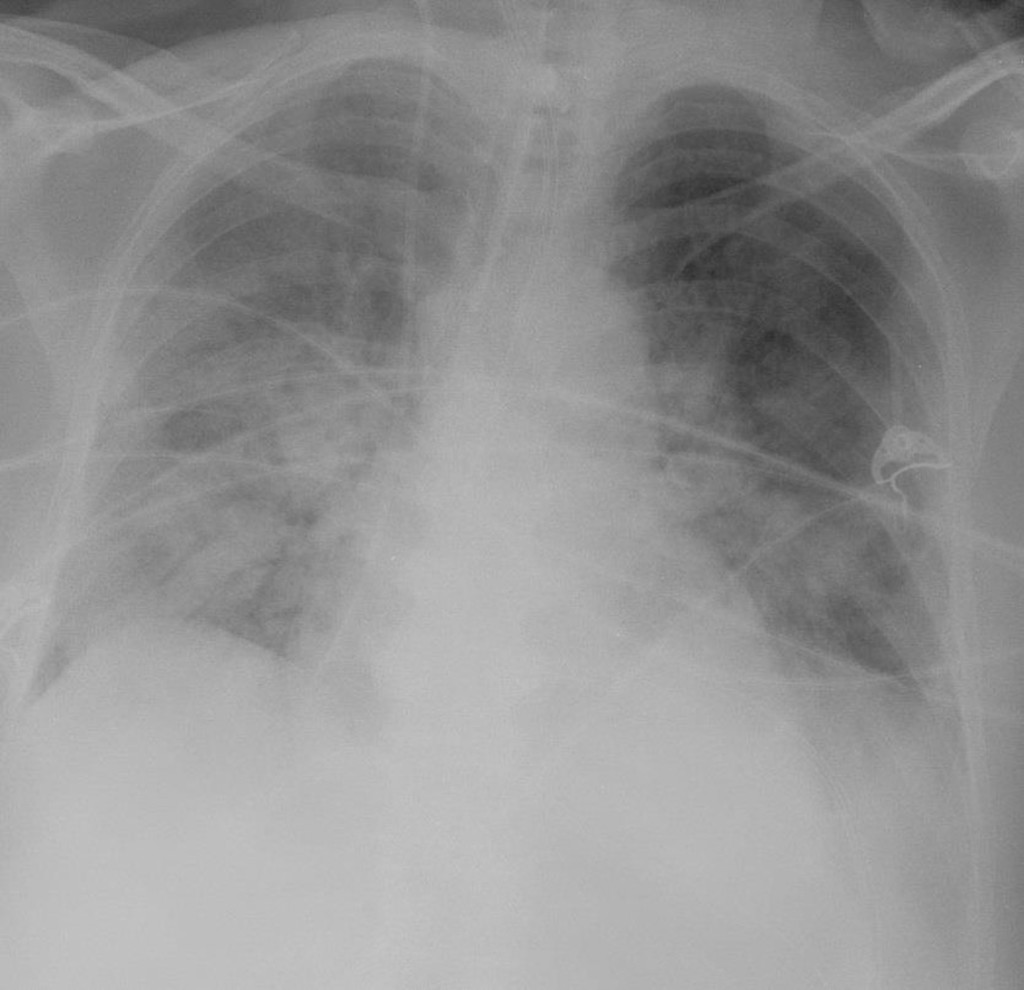
Radiographic findings are an essential part of diagnosing the ARDS patient and will show diffuse bilateral interstitial and airspace densities caused by the edema (figure 5.7). Normal heart and vessel size and absence of pleural effusion distinguish ARDS from cardiogenic pulmonary edema. Although the x-ray gives the appearance of diffuse edema, high-resolution CT often shows that the process is heterogeneous and patchy. This heterogeneity is reflected by remnant patchy fibrosis if the patient recovers; however, the mortality rate for ARDS is around 50 percent.
So now you should have a clear understanding that after an initial insult to the lung an exaggerated and perpetual inflammatory response leads to the destruction of the alveolar–capillary interface. The resulting edema and hyaline membrane formation produces severe hypoxemia and a critically ill patient.
References, Resources, and Further Reading
Text
Farzan, Sattar, with Doris L. Hunsinger and Mary L. Phillips. “Chapter 27.” In A Concise Handbook of Respiratory Diseases. Reston, VA: Reston Publishing Company, 1978.
Husain, Aliya N. “Chapter 15: The Lung.” In Robbins and Cotran Pathologic Basis of Disease, 9th ed., edited by Vinay Kumar, Abul K. Abbas, and John C. Aster. Philadelphia: Saunders, an imprint of Elsevier Inc., 2015.
West, John B. “Chapter 8: Respiratory Failure.” In Pulmonary Pathophysiology: The Essentials, 7th ed. Baltimore: Lippincott Williams & Wilkins, a Wolters Kluwer business, 2008.
Figures
Figure 5.1: The inflammatory response of ARDS causes blockade of pulmonary vasculature. Rosen, Yale. 2010. CC BY-SA 2.0. From Flickr.
Figure 5.2: Pathophysiological events of ARDS. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/5.3_20220203
Figure 5.3: Alveolar wall edema and onset of hyaline membrane formation during the exudative phase of ARDS. Rosen, Yale and Pillappa, Raghavendra. 2009. CC BY-SA 2.0. From Flickr.
Figure 5.4: Proliferation of type II cells and infiltration of fibroblasts in the proliferative phase of ARDS. Rosen, Yale and Pillappa, Raghavendra. 2009. CC BY-SA 2.0. From Flickr.
Figure 5.5: Fibrotic stage of ARDS. Rosen, Yale. 2010. CC BY-SA 2.0. From Flickr.
Figure 5.6: Pathophysiology of ARDS. Grey, Kindred. 2022. CC BY 4.0. https://archive.org/details/5.7_20220203
Figure 5.7: Diffuse bilateral densities associated with ARDS. Lorente, E., et al. 2020. CC BY-NC-SA 3.0. “Image 1” from https://doi.org/10.53347/rID-75182.
