1 Host Defenses, Immunodeficiencies, and Autoimmune Disorders
1.1 Introduction
Despite relatively constant exposure to pathogenic microbes in the environment, humans do not generally suffer from constant infection or disease. Under most circumstances, the body is able to defend itself from the threat of infection thanks to a complex immune system designed to repel, kill, and expel disease-causing invaders. Immunity as a whole can be described as two interrelated parts: nonspecific innate immunity and specific adaptive host defenses.
The nonspecific innate immune response provides a first line of defense that can often prevent infections from gaining a solid foothold in the body. These defenses are described as nonspecific because they do not target any specific pathogen; rather, they defend against a wide range of potential pathogens. They are called innate because they are built-in mechanisms of the human organism. Unlike the specific adaptive defenses, they are not acquired over time and they have no “memory” (they do not improve after repeated exposures to specific pathogens).
Broadly speaking, nonspecific innate defenses provide an immediate (or very rapid) response against potential pathogens. However, these responses are neither perfect nor impenetrable. They can be circumvented by pathogens on occasion, and sometimes they can even cause damage to the body, contributing to the signs and symptoms of infection (figure 1.1).
Nonspecific innate immunity can be characterized as a multifaceted system of defenses that targets invading pathogens in a nonspecific manner. In this chapter, we have divided the numerous defenses that make up this system into three categories: physical defenses, chemical defenses, and cellular defenses. However, it is important to keep in mind that these defenses do not function independently, and the categories often overlap. Table 1.1 provides an overview of the nonspecific defenses discussed in this chapter.
Physical defenses provide the body’s most basic form of nonspecific defense. They include physical barriers to microbes, such as the skin and mucous membranes, as well as mechanical defenses that physically remove microbes and debris from areas of the body where they might cause harm or infection. In addition, the microbiome provides a measure of physical protection against disease, as microbes of the normal microbiota compete with pathogens for nutrients and cellular binding sites necessary to cause infection.
| Overview of nonspecific innate immune defenses | |
|---|---|
| Physical defenses | Physical barriers |
| Mechanical defenses | |
| Microbiome | |
| Chemical defenses | Chemicals and enzymes in body fluids |
| Antimicrobial peptides | |
| Plasma protein mediators | |
| Cytokines | |
| Inflammation-eliciting mediators | |
| Cellular defenses | Granulocytes |
| Agranulocytes | |
Table 1.1: Overview of nonspecific innate immune defenses
1.2 Physical Defenses
Physical Barriers
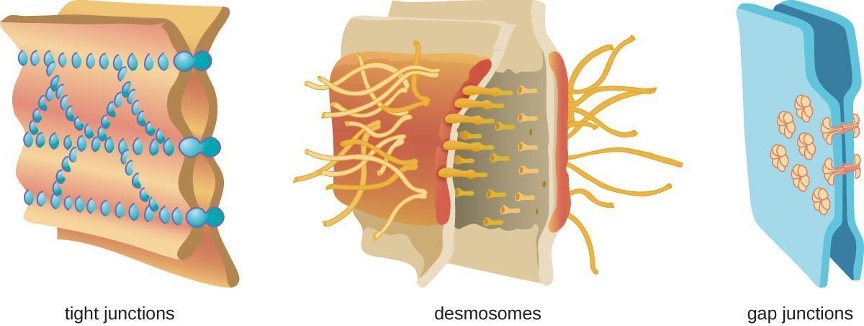
Physical barriers play an important role in preventing microbes from reaching tissues that are susceptible to infection. At the cellular level, barriers consist of cells that are tightly joined to prevent invaders from crossing through to deeper tissue. For example, the endothelial cells that line blood vessels have very tight cell-to-cell junctions, blocking microbes from gaining access to the bloodstream. Cell junctions are generally composed of cell membrane proteins that may connect with the extracellular matrix or with complementary proteins from neighboring cells. Tissues in various parts of the body have different types of cell junctions. These include tight junctions, desmosomes, and gap junctions, as illustrated in figure 1.1. Invading microorganisms may attempt to break down these substances chemically, using enzymes such as proteases that can cause structural damage to create a point of entry for pathogens.
The Skin Barrier
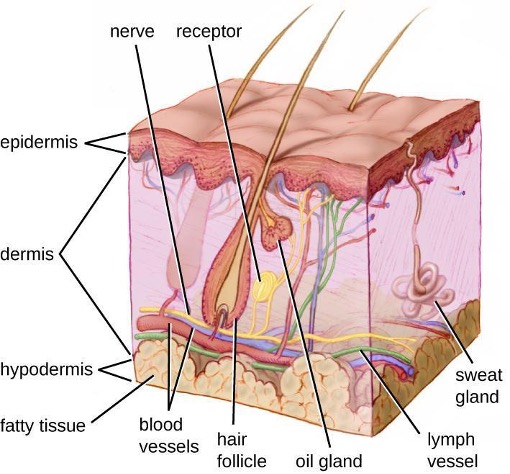
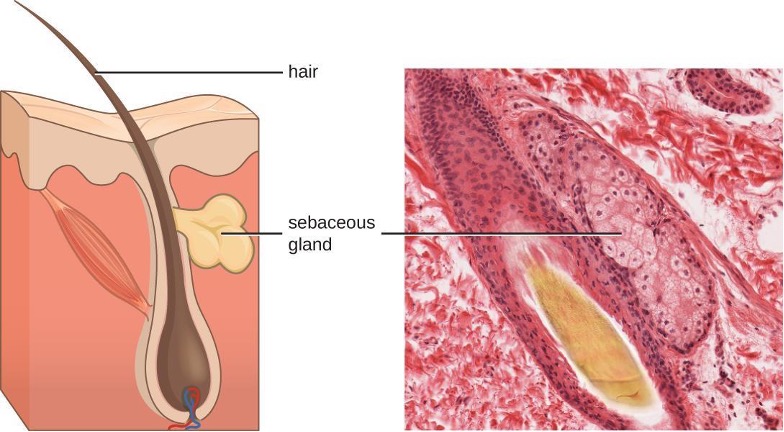
One of the body’s most important physical barriers is the skin barrier, which is composed of three layers of closely packed cells. The thin upper layer is called the epidermis. A second, thicker layer is called the dermis. This layer contains hair follicles, sweat glands, nerves, and blood vessels. A layer of fatty tissue called the hypodermis lies beneath the dermis and contains blood and lymph vessels (figure 1.2).
The topmost layer of skin, the epidermis, consists of cells that are packed with keratin. These dead cells remain as a tightly connected, dense layer of protein-filled cell husks on the surface of the skin. The keratin makes the skin’s surface mechanically tough and resistant to degradation by bacterial enzymes. Fatty acids on the skin’s surface create a dry, salty, and acidic environment that inhibits the growth of some microbes and is highly resistant to breakdown by bacterial enzymes. In addition, the dead cells of the epidermis are frequently shed, along with any microbes that may be clinging to them. Shed skin cells are continually replaced with new cells from below, providing a new barrier that will soon be shed in the same way.
Infections can occur when the skin barrier is compromised or broken. A wound can serve as a point of entry for opportunistic pathogens, which can infect the skin tissue surrounding the wound and possibly spread to deeper tissues.
Mucous Membranes
The mucous membranes lining the nose, mouth, lungs, and urinary and digestive tracts provide another nonspecific barrier against potential pathogens. Mucous membranes consist of a layer of epithelial cells bound by tight junctions. The epithelial cells secrete a moist, sticky substance called mucus, which covers and protects the more fragile cell layers beneath it as well as traps debris and particulate matter, including microbes. Mucus secretions also contain antimicrobial peptides.

In many regions of the body, mechanical actions serve to flush mucus (along with trapped or dead microbes) out of the body or away from potential sites of infection. For example, in the respiratory system, inhalation can bring microbes, dust, mold spores, and other small airborne debris into the body. This debris becomes trapped in the mucus lining the respiratory tract, a layer known as the mucociliary blanket. The epithelial cells lining the upper parts of the respiratory tract are called ciliated epithelial cells because they have hair-like appendages known as cilia. Movement of the cilia propels debris-laden mucus out and away from the lungs. The expelled mucus is then swallowed and destroyed in the stomach, coughed up, or sneezed out (figure 1.3). This system of removal is often called the mucociliary escalator.
The mucociliary escalator is such an effective barrier to microbes that the lungs, the lowermost (and most sensitive) portion of the respiratory tract, were long considered to be a sterile environment in healthy individuals. Only recently has research suggested that healthy lungs may have a small normal microbiota. Disruption of the mucociliary escalator by the damaging effects of smoking or diseases such as cystic fibrosis can lead to increased colonization of bacteria in the lower respiratory tract and frequent infections. These effects highlight the importance of this physical barrier to host defenses.
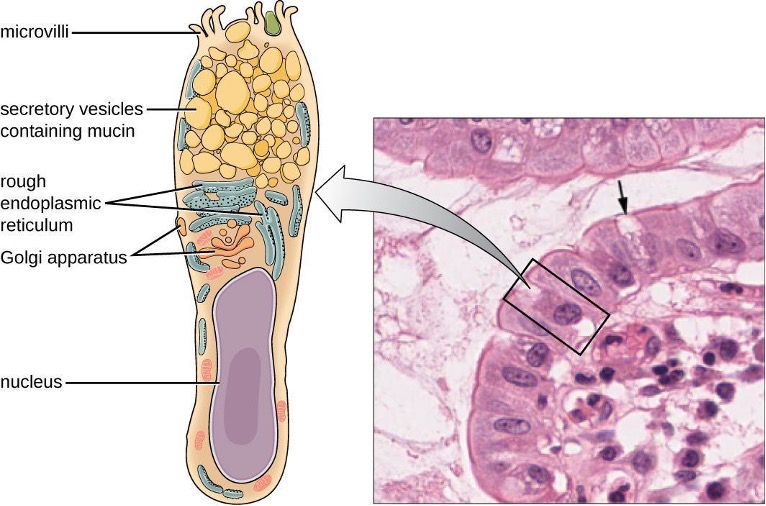
Like the respiratory tract, the digestive tract is a portal of entry through which microbes enter the body, and the mucous membranes lining the digestive tract provide a nonspecific physical barrier against ingested microbes. The intestinal tract is lined with epithelial cells interspersed with mucus-secreting goblet cells (figure 1.4). This mucus mixes with material received from the stomach, trapping foodborne microbes and debris. The mechanical action of peristalsis, a series of muscular contractions in the digestive tract, moves the sloughed mucus and other material through the intestines, rectum, and anus, excreting the material in feces.
Endothelia
The epithelial cells lining the urogenital tract, blood vessels, lymphatic vessels, and certain other tissues are known as endothelia. These tightly packed cells provide a particularly effective frontline barrier against invaders. The endothelia of the blood-brain barrier, for example, protect the central nervous system (CNS), which consists of the brain and the spinal cord. The CNS is one of the most sensitive and important areas of the body, as microbial infection of the CNS can quickly lead to serious and often fatal inflammation. The cell junctions in the blood vessels traveling through the CNS are some of the tightest and toughest in the body, preventing any transient microbes in the bloodstream from entering the CNS. This keeps the cerebrospinal fluid that surrounds and bathes the brain and spinal cord sterile under normal conditions.
1.3 Mechanical Defenses
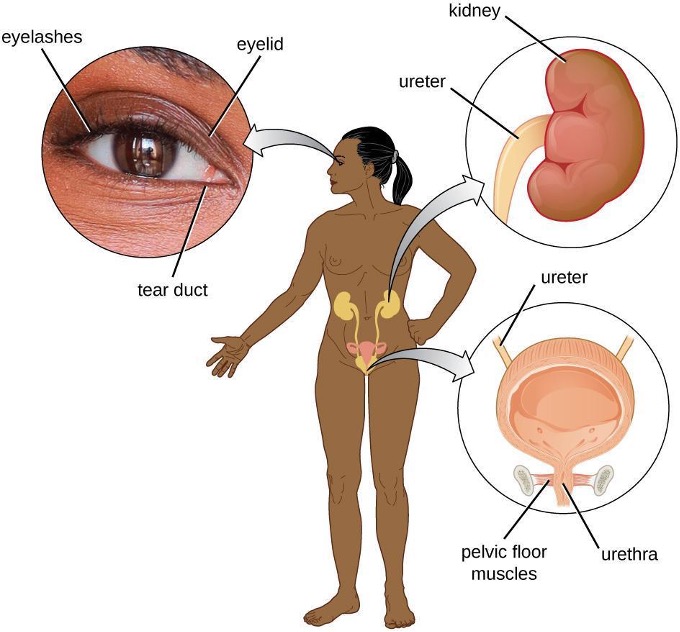
In addition to physical barriers that keep microbes out, the body has a number of mechanical defenses that physically remove pathogens from the body, preventing them from taking up residence. We have already discussed several examples of mechanical defenses, including the shedding of skin cells, the expulsion of mucus via the mucociliary escalator, and the excretion of feces through intestinal peristalsis. Other important examples of mechanical defenses include the flushing action of urine and tears, which both serve to carry microbes away from the body. The flushing action of urine is largely responsible for the normally sterile environment of the urinary tract, which includes the kidneys, ureters, and urinary bladder. Urine passing out of the body washes out transient microorganisms, preventing them from taking up residence. The eyes also have physical barriers and mechanical mechanisms for preventing infections. The eyelashes and eyelids prevent dust and airborne microorganisms from reaching the surface of the eye. Any microbes or debris that make it past these physical barriers may be flushed out by the mechanical action of blinking, which bathes the eye in tears, washing debris away (figure 1.5).
Microbiome
In various regions of the body, resident microbiota serve as an important first-line defense against invading pathogens. Through their occupation of cellular binding sites and competition for available nutrients, the resident microbiota prevent the critical early steps of pathogen attachment and proliferation required for the establishment of an infection. For example, in the vagina, members of the resident microbiota compete with opportunistic pathogens like the yeast Candida. This competition prevents infections by limiting the availability of nutrients, thus inhibiting the growth of Candida, keeping its population in check. Similar competitions occur between the microbiota and potential pathogens on the skin, in the upper respiratory tract, and in the gastrointestinal tract. The resident microbiota also contribute to the chemical defenses of the innate nonspecific host defenses.
The importance of the normal microbiota in host defenses is highlighted by the increased susceptibility to infectious diseases when the microbiota is disrupted or eliminated. Treatment with antibiotics can significantly deplete the normal microbiota of the gastrointestinal tract, providing an advantage for pathogenic bacteria to colonize and cause diarrheal infection. In the case of diarrhea caused by Clostridium difficile, the infection can be severe and potentially lethal. One strategy for treating C. difficile infections is fecal transplantation, which involves the transfer of fecal material from a donor (screened for potential pathogens) into the intestines of the recipient patient as a method of restoring the normal microbiota and combating C. difficile infections.
Table 1.2 provides a summary of the physical defenses discussed in this section.
| Defense | Examples | Function |
|---|---|---|
| Cellular barriers | Skin, mucous membranes, endothelial cells | Deny entry to pathogens |
| Mechanical barriers | Shedding of skin cells, mucociliary sweeping, peristalsis, flushing action of urine and tears | Remove pathogens from potential sites of infection |
| Microbiome | Resident bacteria of the skin, upper respiratory tract, gastrointestinal tract, and genitourinary tract | Compete with pathogens for cellular binding sites and nutrients |
Table 1.2: Physical defenses of nonspecific innate immunity
1.4 Chemical Defenses
In addition to physical defenses, the innate nonspecific immune system uses a number of chemical mediators that inhibit microbial invaders. The term chemical mediators encompasses a wide array of substances found in various body fluids and tissues throughout the body. Chemical mediators may work alone or in conjunction with each other to inhibit microbial colonization and infection.
Some chemical mediators are endogenously produced, meaning they are produced by human body cells; others are produced exogenously, meaning that they are produced by certain microbes that are part of the microbiome. Some mediators are produced continually, bathing the area in the antimicrobial substance; others are produced or activated primarily in response to some stimulus, such as the presence of microbes.
Chemical and Enzymatic Mediators Found in Body Fluids
Fluids produced by the skin include examples of both endogenous and exogenous mediators. Sebaceous glands in the dermis secrete an oil called sebum that is released onto the skin surface through hair follicles. This sebum is an endogenous mediator, providing an additional layer of defense by helping seal off the pore of the hair follicle.This supplemental defense layer prevents bacteria on the skin’s surface from invading sweat glands and surrounding tissue (figure 1.6). Certain members of the microbiome, such as the bacterium Propionibacterium acnes and the fungus Malassezia, among others, can use lipase enzymes to degrade sebum, using it as a food source. This produces oleic acid which creates a mildly acidic environment on the surface of the skin that is inhospitable to many pathogenic microbes. Oleic acid is an example of an exogenously produced mediator because it is produced by resident microbes and not directly by body cells.
Environmental factors that affect the microbiota of the skin can have a direct impact on the production of chemical mediators. Low humidity or decreased sebum production, for example, could make the skin less habitable for microbes that produce oleic acid, thus making the skin more susceptible to pathogens normally inhibited by the skin’s low pH. Many skin moisturizers are formulated to counter such effects by restoring moisture and essential oils to the skin.
The digestive tract also produces a large number of chemical mediators that inhibit or kill microbes. In the oral cavity, saliva contains mediators such as lactoperoxidase enzymes, and mucus secreted by the esophagus contains the antibacterial enzyme lysozyme. In the stomach, highly acidic gastric fluid kills most microbes. In the lower digestive tract, the intestines have pancreatic and intestinal enzymes, antibacterial peptides (cryptins), bile produced from the liver, and specialized Paneth cells that produce lysozyme. Together, these mediators are able to eliminate most pathogens that manage to survive the acidic environment of the stomach.
In the urinary tract, urine flushes microbes out of the body during urination. Furthermore, the slight acidity of urine (the average pH is about 6) inhibits the growth of many microbes and potential pathogens in the urinary tract.
The female reproductive system employs lactate, an exogenously produced chemical mediator, to inhibit microbial growth. The cells and tissue layers composing the vagina produce glycogen, a branched and more complex polymer of glucose. Lactobacilli in the area ferment glycogen to produce lactate, lowering the pH in the vagina thereby inhibiting transient microbiota, opportunistic pathogens like Candida (a yeast associated with vaginal infections), and other pathogens responsible for sexually transmitted diseases.
In the eyes, tears contain the chemical mediators lysozyme and lactoferrin, both of which are capable of eliminating microbes that have found their way to the surface of the eyes. Lysozyme cleaves the bond between NAG and NAM in peptidoglycan, a component of the cell wall in bacteria. It is more effective against gram-positive bacteria, which lack the protective outer membrane associated with gram-negative bacteria. Lactoferrin inhibits microbial growth by chemically binding and sequestering iron. This effectively starves many microbes that require iron for growth.
In the ears, cerumen (earwax) exhibits antimicrobial properties due to the presence of fatty acids, which lower the pH to between 3 and 5.
The respiratory tract uses various chemical mediators in the nasal passages, trachea, and lungs. The mucus produced in the nasal passages contains a mix of antimicrobial molecules similar to those found in tears and saliva (e.g., lysozyme, lactoferrin, lactoperoxidase). Secretions in the trachea and lungs also contain lysozyme and lactoferrin, as well as a diverse group of additional chemical mediators, such as the lipoprotein complex called surfactant, which has antibacterial properties.
Antimicrobial Peptides
The antimicrobial peptides (AMPs) are a special class of nonspecific cell-derived mediators with broad-spectrum antimicrobial properties. Some AMPs are produced routinely by the body, whereas others are primarily produced (or produced in greater quantities) in response to the presence of an invading pathogen. Research has begun exploring how AMPs can be used in the diagnosis and treatment of disease.
AMPs may induce cell damage in microorganisms in a variety of ways, including by inflicting damage to membranes, destroying DNA and RNA, or interfering with cell-wall synthesis. Depending on the specific antimicrobial mechanism, a particular AMP may inhibit only certain groups of microbes (e.g., gram-positive or gram-negative bacteria) or it may be more broadly effective against bacteria, fungi, protozoa, and viruses. Many AMPs are found on the skin, but they can also be found in other regions of the body.
A family of AMPs called defensins can be produced by epithelial cells throughout the body as well as by cellular defenses such as macrophages and neutrophils (see section 1.5). Defensins may be secreted or act inside host cells; they combat microorganisms by damaging their plasma membranes. AMPs called bacteriocins are produced exogenously by certain members of the resident microbiota within the gastrointestinal tract. The genes coding for these types of AMPs are often carried on plasmids and can be passed between different species within the resident microbiota through lateral or horizontal gene transfer.
There are numerous other AMPs throughout the body. The characteristics of a few of the more significant AMPs are summarized in table 1.3.
| AMP | Secreted by | Body site | Pathogens inhibited | Mode of action |
|---|---|---|---|---|
| Bacteriocins | Resident microbiota | Gastrointestinal tract | Bacteria | Disrupt membrane |
| Cathelicidin | Epithelial cells, macrophages, and other cell types | Skin | Bacteria and fungi | Disrupt membrane |
| Defensins | Epithelial cells, macrophages, neutrophils | Throughout the body | Fungi, bacteria, and many viruses | Disrupt membrane |
| Dermcidin | Sweat glands | Skin | Bacteria and fungi | Disrupt membrane integrity and ion channels |
| Histatins | Salivary glands | Oral cavity | Fungi | Disrupt intracellular function |
Table 1.3: Characteristics of selected antimicrobial peptides (AMPs)
Plasma Protein Mediators
Many nonspecific innate immune factors are found in plasma, the fluid portion of blood. Plasma contains electrolytes, sugars, lipids, and proteins, each of which helps to maintain homeostasis (i.e., stable internal body functioning) and contains the proteins involved in the clotting of blood. Additional proteins found in blood plasma, such as acute-phase proteins, complement proteins, and cytokines, are involved in the nonspecific innate immune response.
Acute-Phase Proteins
The acute-phase proteins are another class of antimicrobial mediators. Acute-phase proteins are primarily produced in the liver and secreted into the blood in response to inflammatory molecules from the immune system. Examples of acute-phase proteins include C-reactive protein, serum amyloid A, ferritin, transferrin, fibrinogen, and mannose-binding lectin. Each of these proteins has a different chemical structure and inhibits or destroys microbes in some way (table 1.4).
| Acute-phase protein | Function |
|---|---|
| C-reactive protein | Coats bacteria (opsonization), preparing them for ingestion by phagocytes |
| Serum amyloid A | Can stimulate the secretion of IL-8 from neutrophils |
| Ferritin | Bind and sequester iron, thereby inhibiting the growth of pathogens |
| Transferrin | Ferritin synthesis increases as a nonspecific response aspect of the general pattern of the systemic effects of inflammation ; transferrin is considered a negative acute phase protein |
| Fibrinogen | Involved in formation of blood clots that trap bacterial pathogens |
| Mannose-binding lectin | Activates complement cascade |
Table 1.4: Some acute-phase proteins and their functions
The Complement System
The complement system is a group of plasma protein mediators that can act as an innate nonspecific defense while also serving to connect innate and adaptive immunity. The complement system is composed of more than 30 proteins (including C1 through C9) that normally circulate as precursor proteins in blood. These precursor proteins become activated when triggered by a variety of factors, including the presence of microorganisms. Complement proteins are considered part of innate nonspecific immunity because they are always present in the blood and tissue fluids, allowing them to be activated quickly. Also, when activated through the alternative pathway (described later in this section), complement proteins target pathogens in a nonspecific manner.
The process by which circulating complement precursors become functional is called complement activation. This process is a cascade that can be triggered by one of three different mechanisms, known as the alternative, classical, and lectin pathways.
The alternative pathway is initiated by the spontaneous activation of the complement protein C3. The hydrolysis of C3 produces two products, C3a and C3b. When no invader microbes are present, C3b is very quickly degraded in a hydrolysis reaction using the water in the blood. However, if invading microbes are present, C3b attaches to the surface of these microbes. Once attached, C3b will recruit other complement proteins in a cascade (figure 1.7).
The classical pathway provides a more efficient mechanism of activating the complement cascade, but it depends upon the production of antibodies by the specific adaptive immune defenses. To initiate the classical pathway, a specific antibody must first bind to the pathogen to form an antibody-antigen complex. This activates the first protein in the complement cascade, the C1 complex. The C1 complex is a multipart protein complex, and each component participates in the full activation of the overall complex. Following recruitment and activation of the C1 complex, the remaining classical pathway complement proteins are recruited and activated in a cascading sequence (figure 1.7).
The lectin activation pathway is similar to the classical pathway, but it is triggered by the binding of mannose-binding lectin, an acute-phase protein, to carbohydrates on the microbial surface. Like other acute-phase proteins, lectins are produced by liver cells and are commonly upregulated in response to inflammatory signals received by the body during an infection (figure 1.7).
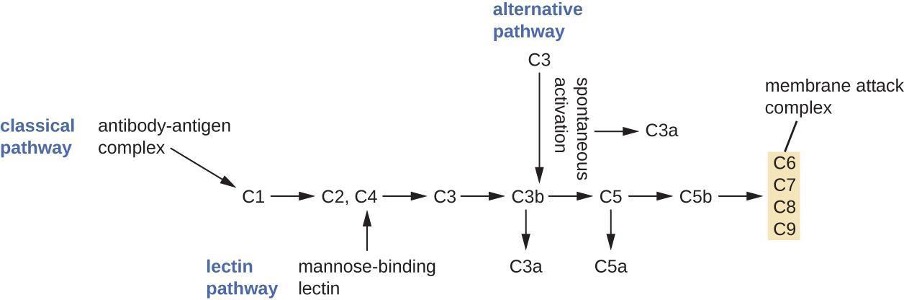
Although each complement activation pathway is initiated in a different way, they all provide the same protective outcomes: opsonization, inflammation, chemotaxis, and cytolysis. The term opsonization refers to the coating of a pathogen by a chemical substance (called an opsonin) that allows phagocytic cells to recognize, engulf, and destroy it more easily. Opsonins from the complement cascade include C1q, C3b, and C4b. Additional important opsonins include mannose-binding proteins and antibodies. The complement fragments C3a and C5a are well-characterized anaphylatoxins with potent proinflammatory functions. Anaphylatoxins activate mast cells, causing degranulation and the release of inflammatory chemical signals, including mediators that cause vasodilation and increased vascular permeability. C5a is also one of the most potent chemoattractants for neutrophils and other white blood cells, cellular defenses that will be discussed in the next section.
The complement proteins C6, C7, C8, and C9 assemble into a membrane attack complex (MAC), which allows C9 to polymerize into pores in the membranes of gram-negative bacteria. These pores allow water, ions, and other molecules to move freely in and out of the targeted cells, eventually leading to cell lysis and death of the pathogen (table 1.5). However, the MAC is only effective against gram-negative bacteria; it cannot penetrate the thick layer of peptidoglycan associated with cell walls of gram-positive bacteria. Since the MAC does not pose a lethal threat to gram-positive bacterial pathogens, complement-mediated opsonization is more important for their clearance.
Cytokines
Cytokines are soluble proteins that act as communication signals between cells. In a nonspecific innate immune response, various cytokines may be released to stimulate production of chemical mediators or other cell functions, such as cell proliferation, cell differentiation, inhibition of cell division, apoptosis, and chemotaxis.
When a cytokine binds to its target receptor, the effect can vary widely depending on the type of cytokine and the type of cell or receptor to which it has bound. The function of a particular cytokine can be described as autocrine, paracrine, or endocrine (table 1.5). In autocrine function, the same cell that releases the cytokine is the recipient of the signal; in other words, autocrine function is a form of self-stimulation by a cell. In contrast, paracrine function involves the release of cytokines from one cell to other nearby cells, stimulating some response from the recipient cells. Last, endocrine function occurs when cells release cytokines into the bloodstream to be carried to target cells much farther away.
| Autocrine | Paracrine | Endocrine |
|---|---|---|
| Same cell secretes and receives cytokine signal. | Cytokine signal secreted to a nearby cell. | Cytokine signal secreted to circulatory system; travels to distant cells. |
 |
 |
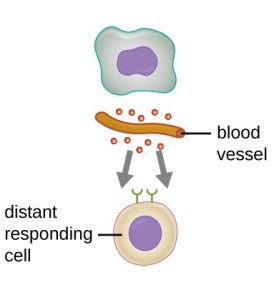 |
Table 1.5: Autocrine, paracrine, and endocrine actions describe which cells are targeted by cytokines and how far the cytokines must travel to bind to their intended target cells’ receptors.
Three important classes of cytokines are the interleukins, chemokines, and interferons. The interleukins were originally thought to be produced only by leukocytes (white blood cells) and to only stimulate leukocytes, thus the reasons for their name. Although interleukins are involved in modulating almost every function of the immune system, their role in the body is not restricted to immunity. Interleukins are also produced by and stimulate a variety of cells unrelated to immune defenses.
The chemokines are chemotactic factors that recruit leukocytes to sites of infection, tissue damage, and inflammation. In contrast to more general chemotactic factors, like complement factor C5a, chemokines are very specific in the subsets of leukocytes they recruit.
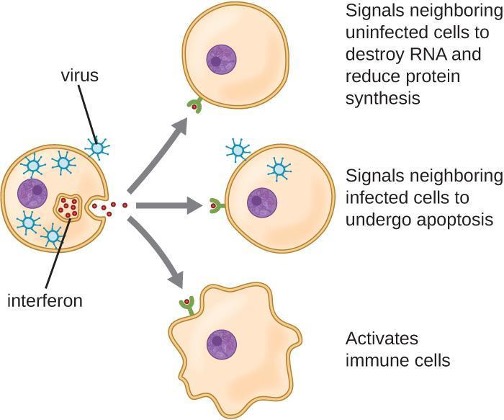
Interferons are a diverse group of immune signaling molecules and are especially important in our defense against viruses. Type I interferons (interferon-α and interferon-β) are produced and released by cells infected with a virus. These interferons stimulate nearby cells to stop production of mRNA, destroy RNA already produced, and reduce protein synthesis. These cellular changes inhibit viral replication and production of mature viruses, slowing the spread of the virus. Type I interferons also stimulate various immune cells involved in viral clearance to more aggressively attack virus-infected cells. Type II interferon (interferon-γ) is an important activator of immune cells (figure 1.8).
Inflammation-Eliciting Mediators
Many of the chemical mediators discussed in this section contribute in some way to inflammation and fever, which are nonspecific immune responses discussed in more detail in section 1.7. Cytokines stimulate the production of acute-phase proteins such as C-reactive protein and mannose-binding lectin in the liver. These acute-phase proteins act as opsonins, activating complement cascades through the lectin pathway.
Some cytokines also bind mast cells and basophils, inducing them to release histamine, a proinflammatory compound. Histamine receptors are found on a variety of cells and mediate proinflammatory events, such as bronchoconstriction (tightening of the airways) and smooth muscle contraction.
In addition to histamine, mast cells may release other chemical mediators, such as leukotrienes. Leukotrienes are lipid-based, proinflammatory mediators that are produced from the metabolism of arachidonic acid in the cell membrane of leukocytes and tissue cells. Compared with the proinflammatory effects of histamine, those of leukotrienes are more potent and longer lasting. Together, these chemical mediators can induce coughing, vomiting, and diarrhea, which serve to expel pathogens from the body.
Certain cytokines also stimulate the production of prostaglandins, chemical mediators that promote the inflammatory effects of kinins and histamines. Prostaglandins can also help to set the body temperature higher, leading to fever, which promotes the activities of white blood cells and slightly inhibits the growth of pathogenic microbes (see section 1.7).
Another inflammatory mediator, bradykinin, contributes to edema, which occurs when fluids and leukocytes leak out of the bloodstream and into tissues. This mediator binds to receptors on cells in the capillary walls, causing the capillaries to dilate and become more permeable to fluids.
Table 1.6 provides a summary of the chemical defenses discussed in this section.
| Defense | Examples | Function |
|---|---|---|
| Chemicals and enzymes in body fluids | Sebum from sebaceous glands | Provides oil barrier protecting hair follicle pores from pathogens |
| Oleic acid from sebum and skin microbiota | Lowers pH to inhibit pathogens | |
| Lysozyme in secretions | Kills bacteria by attacking cell wall | |
| Acid in stomach, urine, and vagina | Inhibits or kills bacteria | |
| Digestive enzymes and bile | Kill bacteria | |
| Lactoferrin and transferrin | Bind and sequester iron, inhibiting bacterial growth | |
| Surfactant in lungs | Kills bacteria | |
| Antimicrobial peptides | Defensins, bacteriocins, dermcidin, cathelicidin, histatins, | Kill bacteria by attacking membranes or interfering with cell functions |
| Plasma protein mediators | Acute-phase proteins (C-reactive protein, serum amyloid A, ferritin, fibrinogen, transferrin, and mannose-binding lectin) | Inhibit the growth of bacteria and assist in the trapping and killing of bacteria |
| Complements C3b and C4b | Opsonization of pathogens to aid phagocytosis | |
| Complement C5a | Chemoattractant for phagocytes | |
| Complements C3a and C5a | Proinflammatory anaphylatoxins | |
| Cytokines | Interleukins | Stimulate and modulate most functions of immune system |
| Chemokines | Recruit white blood cells to infected area | |
| Interferons | Alert cells to viral infection, induce apoptosis of virus-infected cells, induce antiviral defenses in infected and nearby uninfected cells, stimulate immune cells to attack virus-infected cells | |
| Inflammation-eliciting mediators | Histamine | Promotes vasodilation, bronchoconstriction, smooth muscle contraction, increased secretion and mucus production |
| Leukotrienes | Promote inflammation; stronger and longer lasting than histamine | |
| Prostaglandins | Promote inflammation and fever | |
| Bradykinin | Increases vasodilation and vascular permeability, leading to edema |
Table 1.6: Chemical defenses of nonspecific innate immunity
1.5 Cellular Defenses
In the previous section, we discussed some of the chemical mediators found in plasma, the fluid portion of blood. The nonfluid portion of blood consists of various types of formed elements, so called because they are all formed from the same stem cells found in bone marrow. The three major categories of formed elements are: red blood cells (RBCs), also called erythrocytes; platelets, also called thrombocytes; and white blood cells (WBCs), also called leukocytes.
Red blood cells are primarily responsible for carrying oxygen to tissues. Platelets are cellular fragments that participate in blood clot formation and tissue repair. Several different types of WBCs participate in various nonspecific mechanisms of innate and adaptive immunity. In this section, we will focus primarily on the innate mechanisms of various types of WBCs.
Hematopoiesis
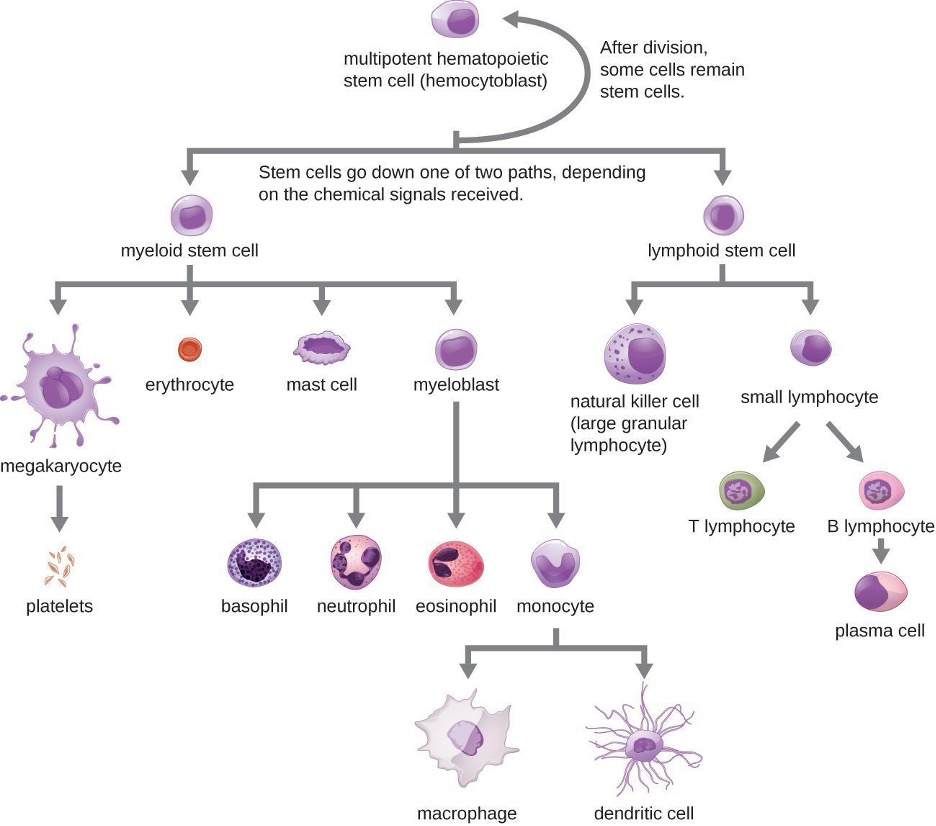
All of the formed elements of blood are derived from pluripotent hematopoietic stem cells (HSCs) in the bone marrow. As the HSCs make copies of themselves in the bone marrow, individual cells receive different cues from the body that control how they develop and mature. As a result, the HSCs differentiate into different types of blood cells that, once mature, circulate in peripheral blood. This process of differentiation, called hematopoiesis, is shown in more detail in figure 1.9.
In terms of sheer numbers, the vast majority of HSCs become erythrocytes. Much smaller numbers become leukocytes and platelets. Leukocytes can be further subdivided into granulocytes, which are characterized by numerous granules visible in the cytoplasm, and agranulocytes, which lack granules. Table 1.7 provides an overview of the various types of formed elements, including their relative numbers, primary function, and lifespans.
| Formed Element | Major Subtypes | Total leukocytes (%) | Numbers Present per Microliter(μL) and Mean (Range) | Appearance in Standard Blood Smear | Summary of Functions | Comments |
|---|---|---|---|---|---|---|
Erythrocytes (red blood cells) |
5.2 million (4.4-6.0 million) |
Flattened biconcave disk; no nucleus; pale red | Transport oxygen and some carbon dioxide between tissue and lungs | Lifespan of approximately 120 days | ||
| Leukocytes (white blood cells) | 7000 (5000-10,000) |
Obvious dark staining nucleus | All Function in body defenses | Exit capillaries and move into tissues; lifespan of usually a few hours or days | ||
| Granulocytes including neutrophils, eosinophils, and basophils | 4360 (1800-9950) |
Abundant granules in cytoplasm, nucleus normally lobed | Nonspecific (innate) resistance to disease | Classified according to membrane-bound granules in cytoplasm | ||
Neutrophils |
50-70 | 4150 (1800-7300) |
Nucleus lobes increase with age; place lilac granules | Phagocytic; particularly effective against bacteria; release cytotoxic chemicals from granules | Most common leukocyte; lifespan of minutes to days | |
Eosinphils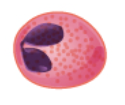 |
1-3 | 165 (0-700) |
Nucleus generally two-lobed; bright red-orange granules | Phagocytic cells; particularly active with antigen-antibody complexes; release degradative enzymes, toxic proteins and antihistamines; combat parasitic infections | lifespan of minutes to days | |
Basophils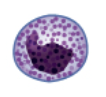 |
<1 | 44 (0-150) |
Nucleus generally two-lobed but difficult to see due to presence of heavy dense , dark purple granules | Pro-inflammatory | Least common leukocyte; lifespan unknown | |
| Agranulocytes including lymphocytes and monocytes | 2640 (1700-4950) |
Lack abundant granules in cytoplasm; have a simple-shaped nucleus that may be indented | Body defenses | Group consists of two major cell types from different linages | ||
Lymphocytes |
20-40 | 2185 (1500-4000) |
Spherical cells with a single, often large, nucleus occupying much of the cell’s volume; stains purple; seen in large (natural killer cells) and small (B and T cells) variants | Primarily specific (adaptive) immunity: T cells directly attack other cells (cellular immunity; B cells release antibodies (humoral immunity); natural killer cells are similar to T cells but nonspecific | Initial cells originate bone marrow, but secondary production occurs in lymphatic tissue; several distinct subtypes; memory cells form after exposure to a pathogen and rapidly increase in response to subsequent exposure; lifespan of many years | |
Monocytes |
1-6 | 455 (200-950) |
Largest leukocyte; has an indented or horseshoe-shaped nucleus | Very effective phagocytic cells engulfing pathogens or worn-out cells; also serve as antigen-presenting cells (APCs) or other components of the immune system | Produced in red bone marrow; referred to as macrophages and dendritic cells after leaving the circulation | |
Platelets |
350,000 (150,000-500,000) |
Cellular fragments surrounded by a plasma membrane and containing granules; stains purple | Hemostasis; release growth factors for repair and healing of tissue | Formed from megakaryocytes that remain in the red bone marrow and shed platelets into circulation |
Table 1.7: Formed elements of blood include erythrocytes (red blood cells), leukocytes (white blood cells), and platelets.
Granulocytes
The various types of granulocytes can be distinguished from one another in a blood smear by the appearance of their nuclei and the contents of their granules, which confer different traits, functions, and staining properties. The neutrophils, also called polymorphonuclear neutrophils (PMNs), have a nucleus with three to five lobes and small, numerous, lilac-colored granules. Each lobe of the nucleus is connected by a thin strand of material to the other lobes. The eosinophils have fewer lobes in the nucleus (typically 2–3) and larger granules that stain reddish-orange. The basophils have a two-lobed nucleus and large granules that stain dark blue or purple (figure 1.10).
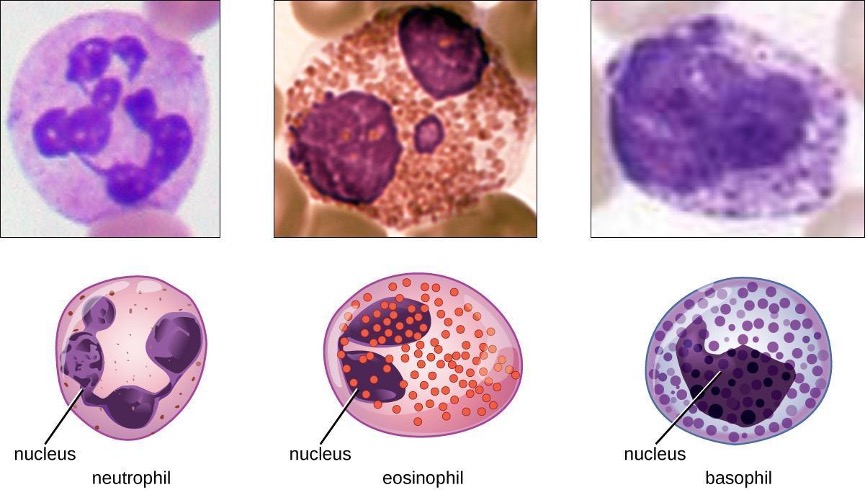
Neutrophils (PMNs)
Neutrophils (PMNs) are frequently involved in the elimination and destruction of extracellular bacteria. They are capable of migrating through the walls of blood vessels to areas of bacterial infection and tissue damage, where they seek out and kill infectious bacteria. PMN granules contain a variety of defensins and hydrolytic enzymes that help them destroy bacteria through phagocytosis (described in more detail in section 1.6). In addition, when many neutrophils are brought into an infected area, they can be stimulated to release toxic molecules into the surrounding tissue to better clear infectious agents. This is called degranulation.
Another mechanism used by neutrophils is neutrophil extracellular traps (NETs), which are extruded meshes of chromatin that are closely associated with antimicrobial granule proteins and components. Chromatin is DNA with associated proteins (usually histone proteins, around which DNA wraps for organization and packing within a cell). By creating and releasing a mesh or lattice-like structure of chromatin that is coupled with antimicrobial proteins, the neutrophils can mount a highly concentrated and efficient attack against nearby pathogens. Proteins frequently associated with NETs include; lactoferrin, gelatinase, cathepsin G, and myeloperoxidase. Each has a different means of promoting antimicrobial activity, helping neutrophils eliminate pathogens. The toxic proteins in NETs may kill some of the body’s own cells along with invading pathogens. However, this collateral damage can be repaired after the danger of the infection has been eliminated.
As neutrophils fight an infection, a visible accumulation of leukocytes, cellular debris, and bacteria at the site of infection can be observed. This buildup is what we call pus (also known as purulent or suppurative discharge or drainage). The presence of pus is a sign that the immune defenses have been activated against an infection; historically, some physicians believed that inducing pus formation could actually promote the healing of wounds. The practice of promoting laudable pus (by, for instance, wrapping a wound in greasy wool soaked in wine) dates back to the ancient physician Galen in the 2nd century AD, and was practiced in variant forms until the 17th century (though it was not universally accepted). Today, this method is no longer practiced because we now know that it is not effective. Although a small amount of pus formation can indicate a strong immune response, artificially inducing pus formation does not promote recovery.
Eosinophils
Eosinophils are granulocytes that protect against protozoa and helminths; they also play a role in allergic reactions. The granules of eosinophils, which readily absorb the acidic reddish dye eosin, contain histamine, degradative enzymes, and a compound known as major basic protein (MBP) (figure 1.10). MBP binds to the surface carbohydrates of parasites, and this binding is associated with disruption of the cell membrane and membrane permeability.
Basophils
Basophils have cytoplasmic granules of varied size and are named for their granules’ ability to absorb the basic dye methylene blue (figure 1.10). Their stimulation and degranulation can result from multiple triggering events. Activated complement fragments C3a and C5a, produced in the activation cascades of complement proteins, act as anaphylatoxins by inducing degranulation of basophils and inflammatory responses. This cell type is important in allergic reactions and other responses that involve inflammation. One of the most abundant components of basophil granules is histamine, which is released along with other chemical factors when the basophil is stimulated. These chemicals can be chemotactic and can help to open the gaps between cells in the blood vessels. Other mechanisms for basophil triggering require the assistance of antibodies, as discussed in section 1.11.
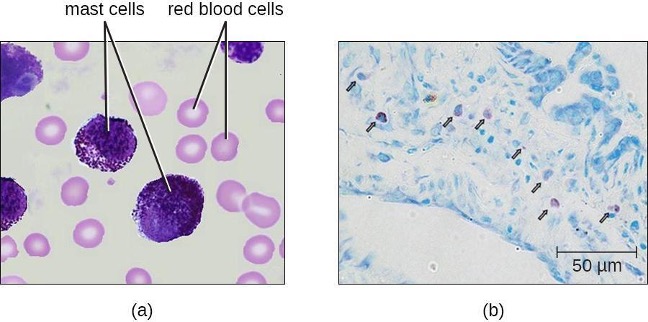
Mast Cells
Hematopoiesis also gives rise to mast cells, which appear to be derived from the same common myeloid progenitor cell as neutrophils, eosinophils, and basophils. Functionally, mast cells are very similar to basophils, containing many of the same components in their granules (e.g., histamine) and playing a similar role in allergic responses and other inflammatory reactions. However, unlike basophils, mast cells leave the circulating blood and are most frequently found residing in tissues. They are often associated with blood vessels and nerves or found close to surfaces that interface with the external environment, such as the skin and mucous membranes in various regions of the body (figure 1.11).
Agranulocytes
As their name suggests, agranulocytes lack visible granules in the cytoplasm. Agranulocytes can be categorized as lymphocytes or monocytes (figure 1.9). Among the lymphocytes are natural killer cells, which play an important role in nonspecific innate immune defenses. Lymphocytes also include the B cells and T cells, and are central players in the specific adaptive immune defenses. The monocytes differentiate into macrophages and dendritic cells, which are collectively referred to as the mononuclear phagocyte system.
Natural Killer Cells
Most lymphocytes are primarily involved in the specific adaptive immune response. An exception is the natural killer cells (NK cells); these mononuclear lymphocytes use nonspecific mechanisms to recognize and destroy cells that are abnormal in some way. Cancer cells and cells infected with viruses are two examples of cellular abnormalities that are targeted by NK cells. Recognition of such cells involves a complex process of identifying inhibitory and activating molecular markers on the surface of the target cell. Molecular markers that make up the major histocompatibility complex (MHC) are expressed by healthy cells as an indication of “self.” NK cells are able to recognize normal MHC markers on the surface of healthy cells, and these MHC markers serve as an inhibitory signal preventing NK cell activation. However, cancer cells and virus-infected cells actively diminish or eliminate expression of MHC markers on their surface. When these MHC markers are diminished or absent, the NK cell interprets this as an abnormality and as a cell in distress. This is one part of the NK cell activation process (figure 1.12). NK cells are also activated by binding to activating molecular molecules on the target cell. These activating molecular molecules include “altered self” or “nonself” molecules. When a NK cell recognizes a decrease in inhibitory normal MHC molecules and an increase in activating molecules on the surface of a cell, the NK cell will be activated to eliminate the cell in distress.
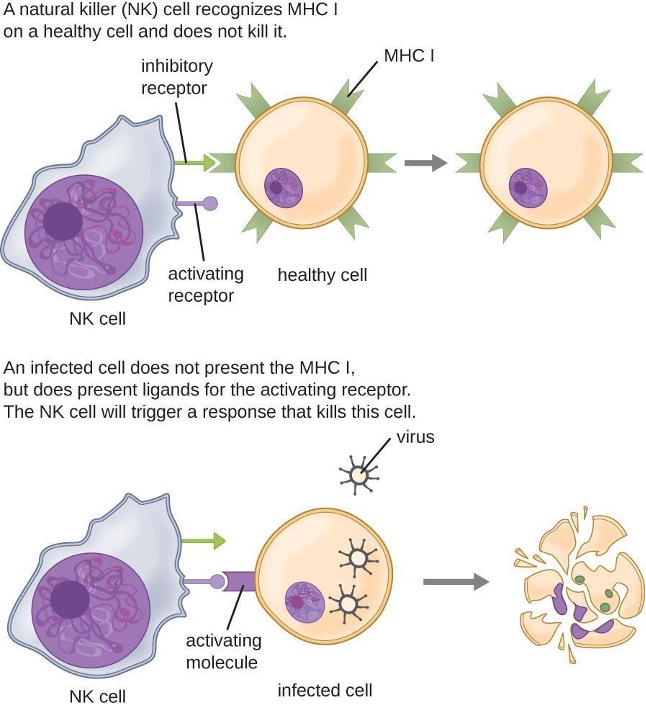
Once a cell has been recognized as a target, the NK cell can use several different mechanisms to kill its target. For example, it may express cytotoxic membrane proteins and cytokines that stimulate the target cell to undergo apoptosis, or controlled cell suicide. NK cells may also use perforin-mediated cytotoxicity to induce apoptosis in target cells. This mechanism relies on two toxins released from granules in the cytoplasm of the NK cell: perforin, a protein that creates pores in the target cell, and granzymes, proteases that enter through the pores into the target cell’s cytoplasm, where they trigger a cascade of protein activation that leads to apoptosis. The NK cell binds to the abnormal target cell, releases its destructive payload, and detaches from the target cell. While the target cell undergoes apoptosis, the NK cell synthesizes more perforin and proteases to use on its next target.
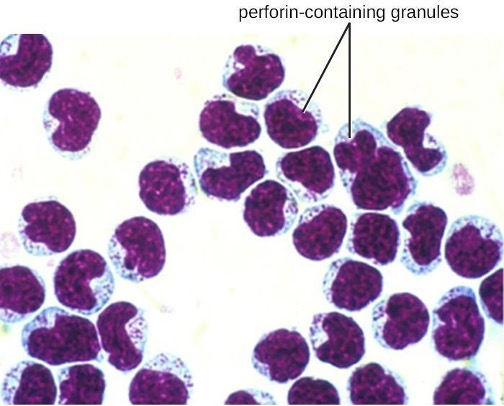
NK cells contain these toxic compounds in granules in their cytoplasm. When stained, the granules are azurophilic and can be visualized under a light microscope (figure 1.13). Even though they have granules, NK cells are not considered granulocytes because their granules are far less numerous than those found in true granulocytes. Furthermore, NK cells have a different lineage than granulocytes, arising from lymphoid rather than myeloid stem cells (figure 1.10).
Monocytes
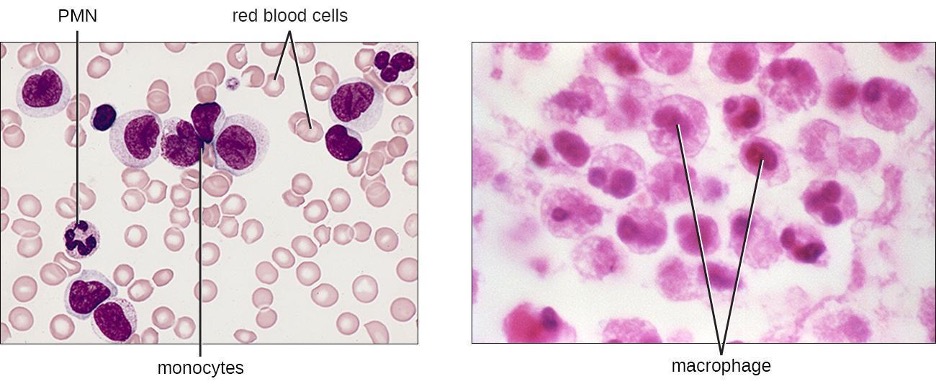
The largest of the white blood cells, monocytes have a nucleus that lacks lobes as well as granules in the cytoplasm (figure 1.14). Nevertheless, they are effective phagocytes, engulfing pathogens and apoptotic cells to help fight infection.
When monocytes leave the bloodstream and enter a specific body tissue, they differentiate into tissue-specific phagocytes called macrophages and dendritic cells. They are particularly important residents of lymphoid tissue, as well as nonlymphoid sites and organs. Macrophages and dendritic cells can reside in body tissues for significant lengths of time. Macrophages in specific body tissues develop characteristics suited to the particular tissue. Not only do they provide immune protection for the tissue in which they reside but they also support normal function of their neighboring tissue cells through the production of cytokines. Macrophages are given tissue-specific names. A few examples of tissue-specific macrophages are listed in table 1.8. Dendritic cells are important sentinels residing in the skin and mucous membranes, which are portals of entry for many pathogens. Monocytes, macrophages, and dendritic cells are all highly phagocytic and are important promoters of the immune response because of their production and release of cytokines. These cells provide an essential bridge between innate and adaptive immune responses.
| Tissue | Macrophage |
|---|---|
| Brain and central nervous system | Microglial cells |
| Liver | Kupffer Cells |
| Lungs | Alveolar macrophages (dust cells) |
| Peritoneal cavity | Peritoneal macrophages |
Table 1.8: Macrophages found in various body tissues
1.6 Pathogen Recognition and Phagocytosis
Several of the cell types discussed in the previous section can be described as phagocytes—cells whose main function is to seek, ingest, and kill pathogens. This process, called phagocytosis, was first observed in starfish in the 1880s by Nobel Prize-winning zoologist Ilya Metchnikoff (1845–1916), who made the connection to white blood cells (WBCs) in humans and other animals. At the time, Louis Pasteur (1822-1895) and other scientists believed that WBCs were spreading pathogens rather than killing them (which is true for some diseases, such as tuberculosis). But in most cases, phagocytes provide a strong, swift, and effective defense against a broad range of microbes, making them a critical component of innate nonspecific immunity. This section will focus on the mechanisms by which phagocytes are able to seek, recognize, and destroy pathogens.
Extravasation (Diapedesis) of Leukocytes
Some phagocytes are leukocytes (WBCs) that normally circulate in the bloodstream. To reach pathogens located in infected tissue, leukocytes must pass through the walls of small capillary blood vessels within tissues. This process, called extravasation, or diapedesis, is initiated by complement factor C5a, as well as cytokines released into the immediate vicinity by resident macrophages and tissue cells responding to the presence of the infectious agent (figure 1.15). Similar to C5a, many of these cytokines are proinflammatory and chemotactic, and they bind to cells of small capillary blood vessels, initiating a response in the endothelial cells lining the inside of the blood vessel walls. This response involves the upregulation and expression of various cellular adhesion molecules and receptors. Leukocytes passing through will stick slightly to the adhesion molecules, slowing down and rolling along the blood vessel walls near the infected area. When they reach a cellular junction, they will bind to even more of these adhesion molecules, flattening out and squeezing through the cellular junction in a process known as transendothelial migration. This mechanism of rolling adhesion allows leukocytes to exit the bloodstream and enter the infected areas, where they can begin phagocytosing the invading pathogens.
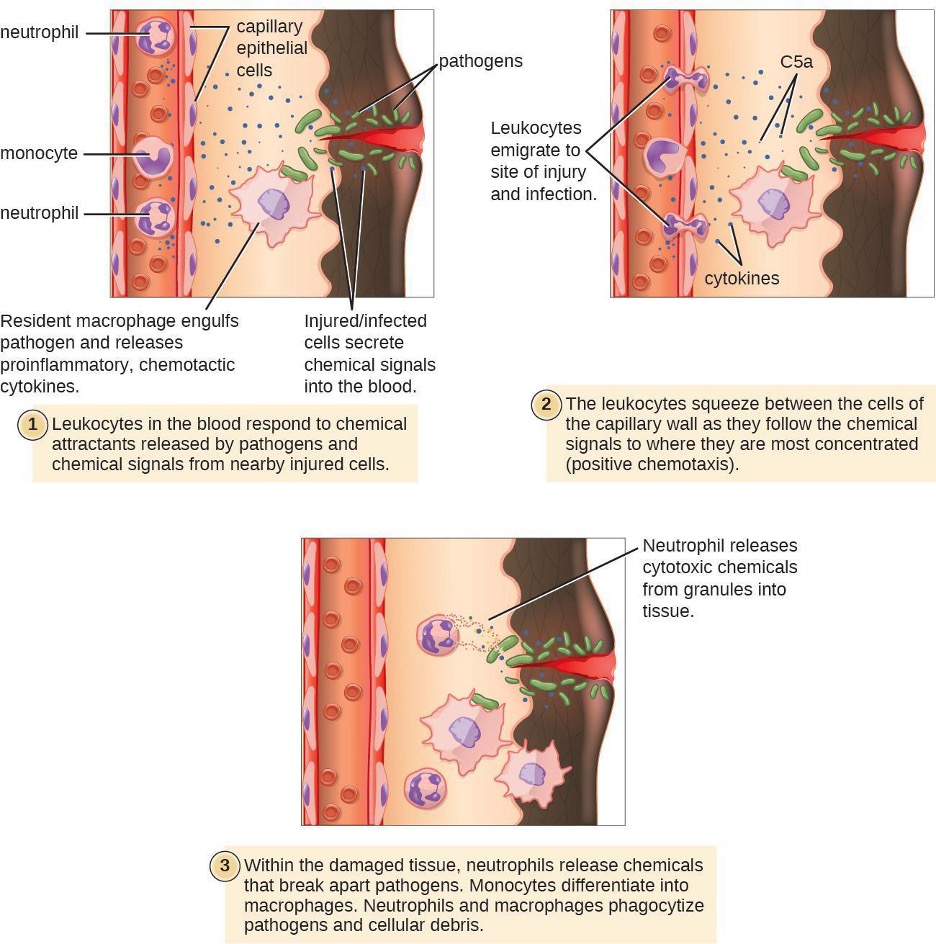
Note that extravasation does not occur in arteries or veins. These blood vessels are surrounded by thicker, multilayer protective walls, in contrast to the thin single-cell-layer walls of capillaries. Furthermore, the blood flow in arteries is too turbulent to allow for rolling adhesion. Also, some leukocytes tend to respond to an infection more quickly than others. The first to arrive typically are neutrophils, often within hours of a bacterial infection. By contract, monocytes may take several days to leave the bloodstream and differentiate into macrophages.
Pathogen Recognition
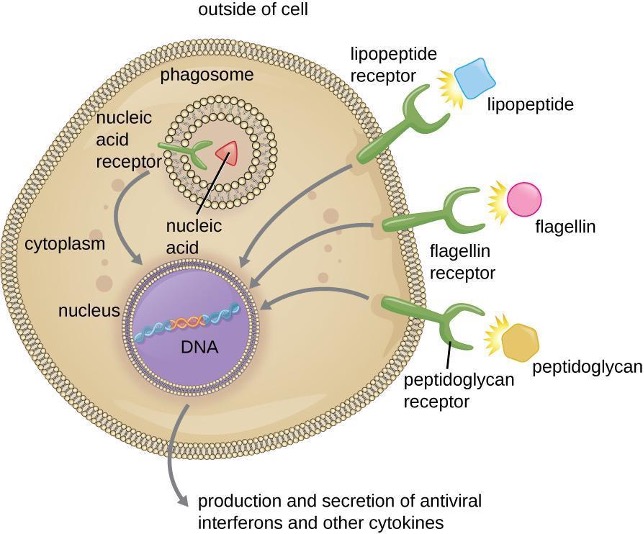
As described in the previous section, opsonization of pathogens by antibody; complement factors C1q, C3b, and C4b; and lectins can assist phagocytic cells in recognition of pathogens and attachment to initiate phagocytosis. However, not all pathogen recognition is opsonin dependent. Phagocytes can also recognize molecular structures that are common to many groups of pathogenic microbes. Such structures are called pathogen-associated molecular patterns (PAMPs). Common PAMPs include the following:
- peptidoglycan, found in bacterial cell walls;
- flagellin, a protein found in bacterial flagella;
- lipopolysaccharide (LPS) from the outer membrane of gram-negative bacteria;
- lipopeptides, molecules expressed by most bacteria; and
- nucleic acids such as viral DNA or RNA.
Like numerous other PAMPs, these substances are integral to the structure of broad classes of microbes.
The structures that allow phagocytic cells to detect PAMPs are called pattern recognition receptors (PRRs). One group of PRRs is the toll-like receptors (TLRs), which bind to various PAMPs and communicate with the nucleus of the phagocyte to elicit a response. Many TLRs (and other PRRs) are located on the surface of a phagocyte, but some can also be found embedded in the membranes of interior compartments and organelles (figure 1.16). These interior PRRs can be useful for the binding and recognition of intracellular pathogens that may have gained access to the inside of the cell before phagocytosis could take place. Viral nucleic acids, for example, might encounter an interior PRR, triggering production of the antiviral cytokine interferon.
In addition to providing the first step of pathogen recognition, the interaction between PAMPs and PRRs on macrophages provides an intracellular signal that activates the phagocyte, causing it to transition from a dormant state of readiness and slow proliferation to a state of hyperactivity, proliferation, production/secretion of cytokines, and enhanced intracellular killing. PRRs on macrophages also respond to chemical distress signals from damaged or stressed cells. This allows macrophages to extend their responses beyond protection from infectious diseases to a broader role in the inflammatory response initiated from injuries or other diseases.
Pathogen Degradation
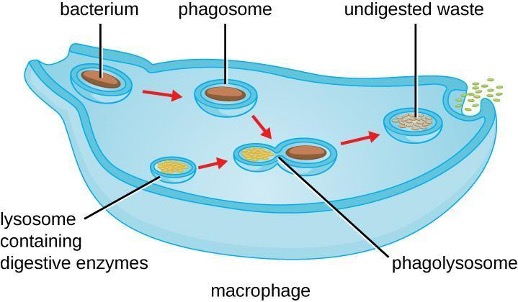
Once pathogen recognition and attachment occurs, the pathogen is engulfed in a vesicle and brought into the internal compartment of the phagocyte in a process called phagocytosis (figure 1.17). PRRs can aid in phagocytosis by first binding to the pathogen’s surface, but phagocytes are also capable of engulfing nearby items even if they are not bound to specific receptors. To engulf the pathogen, the phagocyte forms a pseudopod that wraps around the pathogen and then pinches it off into a membrane vesicle called a phagosome. Acidification of the phagosome (pH decreases to the range of 4–5) provides an important early antibacterial mechanism. The phagosome containing the pathogen fuses with one or more lysosomes, forming a phagolysosome. Formation of the phagolysosome enhances the acidification, which is essential for activation of pH-dependent digestive lysosomal enzymes and production of hydrogen peroxide and toxic reactive oxygen species. Lysosomal enzymes such as lysozyme, phospholipase, and proteases digest the pathogen. Other enzymes are involved in a respiratory burst. During the respiratory burst, phagocytes will increase their uptake and consumption of oxygen, but not for energy production. The increased oxygen consumption is focused on the production of superoxide anion, hydrogen peroxide, hydroxyl radicals, and other reactive oxygen species that are antibacterial.
In addition to the reactive oxygen species produced by the respiratory burst, reactive nitrogen compounds with cytotoxic (cell-killing) potential can also form. For example, nitric oxide can react with superoxide to form peroxynitrite, a highly reactive nitrogen compound with degrading capabilities similar to those of the reactive oxygen species. Some phagocytes even contain an internal storehouse of microbicidal defensin proteins (e.g., neutrophil granules). These destructive forces can be released into the area around the cell to degrade microbes externally. Neutrophils, especially, can be quite efficient at this secondary antimicrobial mechanism.
Once degradation is complete, leftover waste products are excreted from the cell in an exocytic vesicle. However, it is important to note that not all remains of the pathogen are excreted as waste. Macrophages and dendritic cells are also antigen-presenting cells involved in the specific adaptive immune response. These cells further process the remains of the degraded pathogen and present key antigens (specific pathogen proteins) on their cellular surface. This is an important step for stimulation of some adaptive immune responses.
1.7 Inflammation and Fever
The inflammatory response, or inflammation, is triggered by a cascade of chemical mediators and cellular responses that may occur when cells are damaged and stressed or when pathogens successfully breach the physical barriers of the innate immune system. Although inflammation is typically associated with negative consequences of injury or disease, it is a necessary process insofar as it allows for recruitment of the cellular defenses needed to eliminate pathogens, remove damaged and dead cells, and initiate repair mechanisms. Excessive inflammation, however, can result in local tissue damage and, in severe cases, may even become deadly.
Acute Inflammation
An early, if not immediate, response to tissue injury is acute inflammation. Immediately following an injury, vasoconstriction of blood vessels will occur to minimize blood loss. The amount of vasoconstriction is related to the amount of vascular injury, but it is usually brief. Vasoconstriction is followed by vasodilation and increased vascular permeability, as a direct result of the release of histamine from resident mast cells. Increased blood flow and vascular permeability can dilute toxins and bacterial products at the site of injury or infection. They also contribute to the five observable signs associated with the inflammatory response: erythema (redness), edema (swelling), heat, pain, and altered function. Vasodilation and increased vascular permeability are also associated with an influx of phagocytes at the site of injury and/or infection. This can enhance the inflammatory response because phagocytes may release proinflammatory chemicals when they are activated by cellular distress signals released from damaged cells, by PAMPs, or by opsonins on the surface of pathogens. Activation of the complement system can further enhance the inflammatory response through the production of the anaphylatoxin C5a. Figure 1.18 illustrates a typical case of acute inflammation at the site of a skin wound.
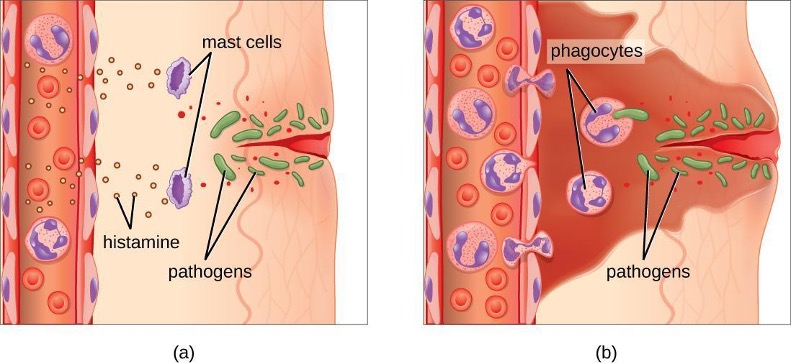
During the period of inflammation, the release of bradykinin causes capillaries to remain dilated, flooding tissues with fluids and leading to edema. Increasing numbers of neutrophils are recruited to the area to fight pathogens. As the fight rages on, pus forms from the accumulation of neutrophils, dead cells, tissue fluids, and lymph. Typically, after a few days, macrophages will help to clear out this pus. Eventually, tissue repair can begin in the wounded area.
Chronic Inflammation
When acute inflammation is unable to clear an infectious pathogen, chronic inflammation may occur. This often results in an ongoing (and sometimes futile) lower-level battle between the host organism and the pathogen. The wounded area may heal at a superficial level, but pathogens may still be present in deeper tissues, stimulating ongoing inflammation. Additionally, chronic inflammation may be involved in the progression of degenerative neurological diseases such as Alzheimer’s, Parkinson’s, heart disease, and metastatic cancer.
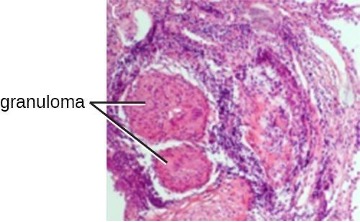
Chronic inflammation may lead to the formation of granulomas, pockets of infected tissue walled off and surrounded by WBCs. Macrophages and other phagocytes wage an unsuccessful battle to eliminate the pathogens and dead cellular materials within a granuloma. One example of a disease that produces chronic inflammation is tuberculosis, which results in the formation of granulomas in lung tissues. A tubercular granuloma is called a tubercle (figure 1.19). Tuberculosis will be covered in more detail in section 5.2.
Chronic inflammation is not just associated with bacterial infections. Chronic inflammation can be an important cause of tissue damage from viral infections. The extensive scarring observed with hepatitis C infections and liver cirrhosis is the result of chronic inflammation.
Fever
A fever is an inflammatory response that extends beyond the site of infection and affects the entire body, resulting in an overall increase in body temperature. Body temperature is normally regulated and maintained by the hypothalamus, an anatomical section of the brain that functions to maintain homeostasis in the body. However, certain bacterial or viral infections can result in the production of pyrogens, chemicals that effectively alter the “thermostat setting” of the hypothalamus to elevate body temperature and cause fever. Pyrogens may be exogenous or endogenous. For example, the endotoxin lipopolysaccharide (LPS), produced by gram-negative bacteria, is an exogenous pyrogen that may induce the leukocytes to release endogenous pyrogens such as interleukin-1 (IL-1), IL-6, interferon-γ (IFN-γ), and tumor necrosis factor (TNF). In a cascading effect, these molecules can then lead to the release of prostaglandin E2 (PGE2) from other cells, resetting the hypothalamus to initiate fever (figure 1.20).
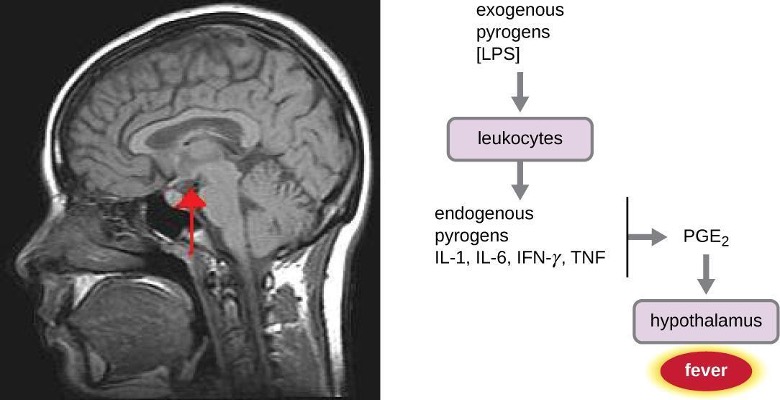
Like other forms of inflammation, a fever enhances the innate immune defenses by stimulating leukocytes to kill pathogens. The rise in body temperature also may inhibit the growth of many pathogens since human pathogens are mesophiles with optimum growth occurring around 35 °C (95 °F). In addition, some studies suggest that fever may also stimulate release of iron-sequestering compounds from the liver, thereby starving out microbes that rely on iron for growth.[1]
During fever, the skin may appear pale due to vasoconstriction of the blood vessels in the skin, which is mediated by the hypothalamus to divert blood flow away from extremities, minimizing the loss of heat and raising the core temperature. The hypothalamus will also stimulate shivering of muscles, another effective mechanism of generating heat and raising the core temperature.
The crisis phase occurs when the fever breaks. The hypothalamus stimulates vasodilation, resulting in a return of blood flow to the skin and a subsequent release of heat from the body. The hypothalamus also stimulates sweating, which cools the skin as the sweat evaporates.
Although a low-level fever may help an individual overcome an illness, in some instances, this immune response can be too strong, causing tissue and organ damage and, in severe cases, even death. The inflammatory response to bacterial superantigens is one scenario in which a life-threatening fever may develop. Superantigens are bacterial or viral proteins that can cause an excessive activation of T cells from the specific adaptive immune defense, as well as an excessive release of cytokines that overstimulates the inflammatory response. For example, Staphylococcus aureus and Streptococcus pyogenes are capable of producing superantigens that cause toxic shock syndrome and scarlet fever, respectively. Both of these conditions can be associated with very high, life-threatening fevers in excess of 42 °C (108 °F).
1.8 Overview of Specific Adaptive Immunity
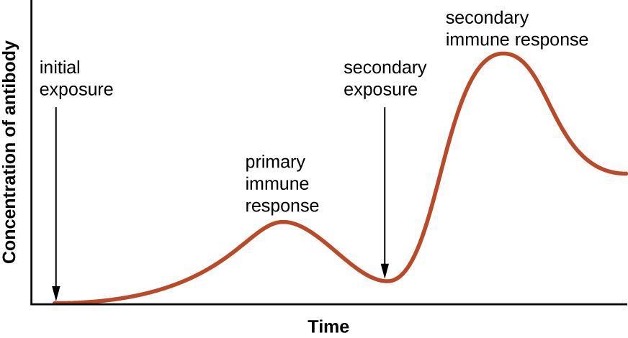
Adaptive immunity is defined by two important characteristics: specificity and memory. Specificity refers to the adaptive immune system’s ability to target specific pathogens, and memory refers to its ability to quickly respond to pathogens to which it has previously been exposed. For example, when an individual recovers from chickenpox, the body develops a memory of the infection that will specifically protect it from the causative agent, the varicella-zoster virus, if it is exposed to the virus again.
Specificity and memory are achieved by essentially programming certain cells involved in the immune response to respond rapidly to subsequent exposures of the pathogen. This programming occurs as a result of the first exposure to a pathogen or vaccine, which triggers a primary response. Subsequent exposures result in a secondary response that is faster and stronger as a result of the body’s memory of the first exposure (figure 1.21). This secondary response, however, is specific to the pathogen in question. For example, exposure to one virus (e.g., varicella-zoster virus) will not provide protection against other viral diseases (e.g., measles, mumps, or polio).
Adaptive specific immunity involves the actions of two distinct cell types: B lymphocytes (B cells) and T lymphocytes (T cells). Although B cells and T cells arise from a common hematopoietic stem cell differentiation pathway (see figure 1.9), their sites of maturation and their roles in adaptive immunity are very different.
B cells mature in the bone marrow and are responsible for the production of glycoproteins called antibodies, or immunoglobulins. Antibodies are involved in the body’s defense against pathogens and toxins in the extracellular environment. Mechanisms of adaptive specific immunity that involve B cells and antibody production are referred to as humoral immunity. The maturation of T cells occurs in the thymus. T cells function as the central orchestrator of both innate and adaptive immune responses. They are also responsible for destruction of cells infected with intracellular pathogens. The targeting and destruction of intracellular pathogens by T cells is called cell-mediated immunity, or cellular immunity.
Antigens
Activation of the adaptive immune defenses is triggered by pathogen-specific molecular structures called antigens. Antigens are similar to the pathogen-associated molecular patterns (PAMPs) discussed in section 1.6; however, whereas PAMPs are molecular structures found on numerous pathogens, antigens are unique to a specific pathogen. The antigens that stimulate adaptive immunity to chickenpox, for example, are unique to the varicella-zoster virus but significantly different from the antigens associated with other viral pathogens.
The term antigen was initially used to describe molecules that stimulate the production of antibodies; in fact, the term comes from a combination of the words antibody and generator, and a molecule that stimulates antibody production is said to be antigenic. However, the role of antigens is not limited to humoral immunity and the production of antibodies; antigens also play an essential role in stimulating cellular immunity, and for this reason antigens are sometimes more accurately referred to as immunogens. In this text, however, we will typically refer to them as antigens.
Pathogens possess a variety of structures that may contain antigens. For example, antigens from bacterial cells may be associated with their capsules, cell walls, fimbriae, flagella, or pili. Bacterial antigens may also be associated with extracellular toxins and enzymes that they secrete. Viruses possess a variety of antigens associated with their capsids, envelopes, and the spike structures they use for attachment to cells.
Antigens may belong to any number of molecular classes, including carbohydrates, lipids, nucleic acids, proteins, and combinations of these molecules. Antigens of different classes vary in their ability to stimulate adaptive immune defenses as well as in the type of response they stimulate (humoral or cellular). The structural complexity of an antigenic molecule is an important factor in its antigenic potential. In general, more complex molecules are more effective as antigens. For example, the three-dimensional complex structure of proteins make them the most effective and potent antigens, capable of stimulating both humoral and cellular immunity. In comparison, carbohydrates are less complex in structure and therefore less effective as antigens; they can only stimulate humoral immune defenses. Lipids and nucleic acids are the least antigenic molecules. In some cases these structures may only become antigenic when combined with proteins or carbohydrates to form glycolipids, lipoproteins, or nucleoproteins.
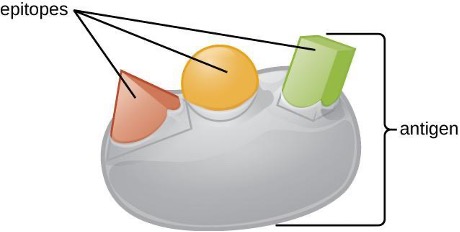
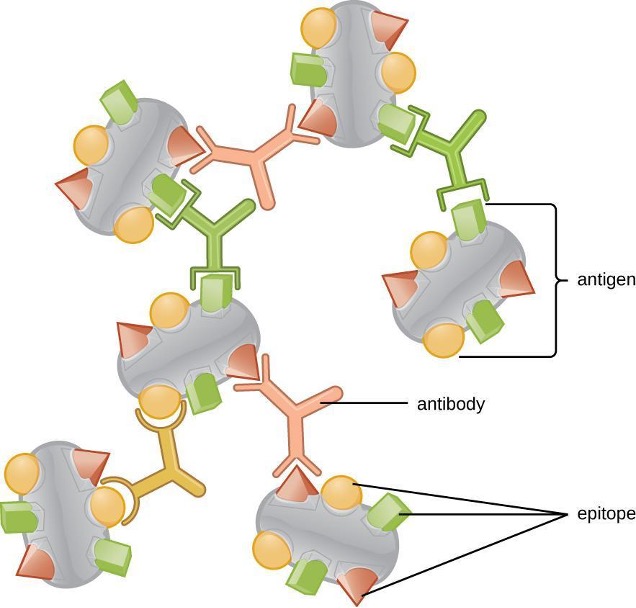
One reason the three-dimensional complexity of antigens is so important is that antibodies and T cells do not recognize and interact with an entire antigen but with smaller exposed regions on the surface of antigens called epitopes. A single antigen may possess several different epitopes (figure 1.22), and different antibodies may bind to different epitopes on the same antigen (figure 1.23). For example, the bacterial flagellum is a large, complex protein structure that can possess hundreds or even thousands of epitopes with unique three-dimensional structures. Moreover, flagella from different bacterial species (or even strains of the same species) contain unique epitopes that can only be bound by specific antibodies.
An antigen’s size is another important factor in its antigenic potential. Whereas large antigenic structures like flagella possess multiple epitopes, some molecules are too small to be antigenic by themselves. Such molecules, called haptens, are essentially free epitopes that are not part of the complex three-dimensional structure of a larger antigen. For a hapten to become antigenic, it must first attach to a larger carrier molecule (usually a protein) to produce a conjugate antigen. The hapten-specific antibodies produced in response to the conjugate antigen are then able to interact with unconjugated free hapten molecules. Haptens are not known to be associated with any specific pathogens, but they are responsible for some allergic responses. For example, the hapten urushiol, a molecule found in the oil of plants that cause poison ivy, causes an immune response that can result in a severe rash (called contact dermatitis). Similarly, the hapten penicillin can cause allergic reactions to drugs in the penicillin class.
Antibodies
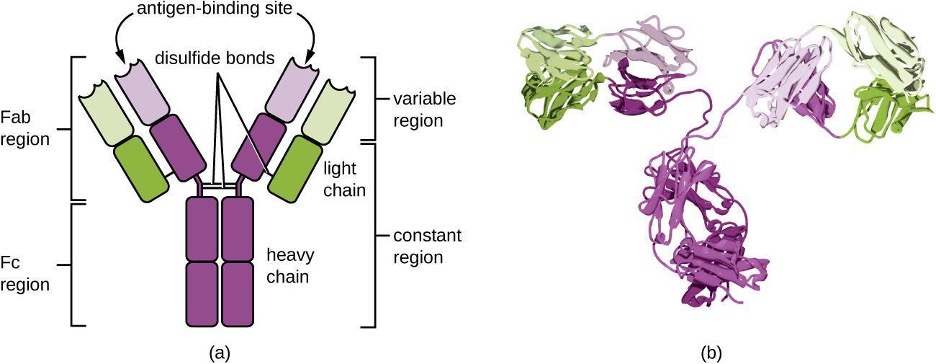
Antibodies (also called immunoglobulins) are glycoproteins that are present in both the blood and tissue fluids. The basic structure of an antibody monomer consists of four protein chains held together by disulfide bonds (figure 1.24). A disulfide bond is a covalent bond between the sulfhydryl R groups found on two cysteine amino acids. The two largest chains are identical to each other and are called the heavy chains. The two smaller chains are also identical to each other and are called the light chains. Joined together, the heavy and light chains form a basic Y-shaped structure.
The two “arms” of the Y-shaped antibody molecule are known as the Fab region (fragment of antigen binding). The far end of the Fab region is the variable region, which serves as the site of antigen binding. The amino acid sequence in the variable region dictates the three-dimensional structure, and thus the specific three-dimensional epitope to which the Fab region is capable of binding. Although the epitope specificity of the Fab regions is identical for each arm of a single antibody molecule, this region displays a high degree of variability between antibodies with different epitope specificities. Binding to the Fab region is necessary for neutralization of pathogens, agglutination or aggregation of pathogens, and antibody-dependent cell-mediated cytotoxicity.
The constant region of the antibody molecule includes the trunk of the Y and lower portion of each arm of the Y. The trunk of the Y is also called the Fc region (fragment of crystallization) and is the site of complement factor binding and binding to phagocytic cells during antibody-mediated opsonization.
Antibody Classes
The constant region of an antibody molecule determines its class, or isotype. The five classes of antibodies are IgG, IgM, IgA, IgD, and IgE. Each class possesses unique heavy chains designated by the Greek letters γ, μ, α, δ, and ε, respectively. Antibody classes also exhibit important differences in abundance in serum, arrangement, body sites of action, functional roles, and size (table 1.9).
IgG is a monomer that is by far the most abundant antibody in human blood, accounting for about 80% of total serum antibody. IgG penetrates efficiently into tissue spaces and is the only antibody class with the ability to cross the placental barrier, providing passive immunity to the developing fetus during pregnancy. IgG is also the most versatile antibody class in terms of its role in the body’s defense against pathogens.
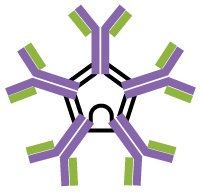
IgM is initially produced in a monomeric membrane-bound form that serves as an antigen-binding receptor on B cells. The secreted form of IgM assembles into a pentamer with five monomers of IgM bound together by a protein structure called the J chain. Although the location of the J chain relative to the Fc regions of the five monomers prevents IgM from performing some of the functions of IgG, the ten available Fab sites associated with a pentameric IgM make it an important antibody in the body’s arsenal of defenses. IgM is the first antibody produced and secreted by B cells during the primary and secondary immune responses, making pathogen-specific IgM a valuable diagnostic marker during active or recent infections.
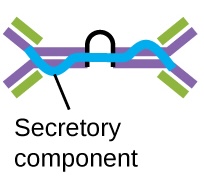
IgA accounts for about 13% of total serum antibody, and secretory IgA is the most common and abundant antibody class found in the mucus secretions that protect the mucous membranes. IgA can also be found in other secretions such as breast milk, tears, and saliva. Secretory IgA is assembled into a dimeric form with two monomers joined by a protein structure called the secretory component. One of the important functions of secretory IgA is to trap pathogens in mucus so that they can later be eliminated from the body.
Similar to IgM, IgD is a membrane-bound monomer found on the surface of B cells, where it serves as an antigen-binding receptor. However, IgD is not secreted by B cells, and only trace amounts are detected in serum. These trace amounts most likely come from the degradation of old B cells and the release of IgD molecules from their cytoplasmic membranes.
IgE is the least abundant antibody class in serum. Like IgG, it is secreted as a monomer, but its role in adaptive immunity is restricted to anti-parasitic defenses. The Fc region of IgE binds to basophils and mast cells. The Fab region of the bound IgE then interacts with specific antigen epitopes, causing the cells to release potent pro-inflammatory mediators. The inflammatory reaction resulting from the activation of mast cells and basophils aids in the defense against parasites, but this reaction is also central to allergic reactions.
| Properties | IgG monomer | IgM pentamer | Secretory IgA dimer | IgD monomer | IgE Monomer |
|---|---|---|---|---|---|
| Structure | 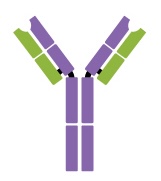 |
 |
 |
|
|
| Heavy chains | γ | μ | α | δ | ε |
| Number of antigen-binding sites | 2 | 10 | 4 | 2 | 2 |
| Molecular weight (Daltons) | 150,000 | 900,000 | 385,000 | 180,000 | 200,000 |
| Percentage of total antibody in serum | 80% | 6% | 13% (monomer) | <1% | <1% |
| Crosses placenta | yes | no | no | no | no |
| Fixes complement | yes | yes | no | no | no |
| Fc binds to | phagocytes | mast cells and basophils | |||
| Function | Neutralization, agglutination, complement activation, opsonization, and antibody-dependent cell-mediated cytotoxicity. | Neutralization, agglutination, complement activation. The monomer serves as the B-cell receptor. | Neutralization and trapping of pathogens in mucus. | B-cell receptor. | Activation of basophils and mast cells against parasites and allergens. |
Table 1.9: The five immunoglobulin (Ig) classes
Antigen-Antibody Interactions
Different classes of antibodies play important roles in the body’s defense against pathogens. These functions include neutralization of pathogens, opsonization for phagocytosis, agglutination, complement activation, and antibody-dependent cell-mediated cytotoxicity. For most of these functions, antibodies also provide an important link between adaptive specific immunity and innate nonspecific immunity.
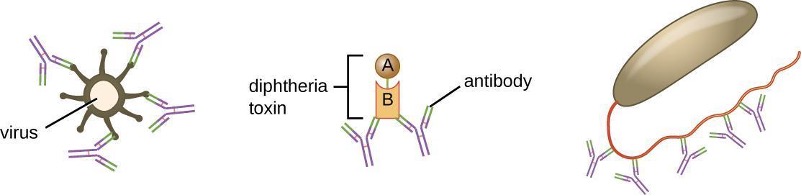
Neutralization involves the binding of certain antibodies (IgG, IgM, or IgA) to epitopes on the surface of pathogens or toxins, preventing their attachment to cells. For example, Secretory IgA can bind to specific pathogens and block initial attachment to intestinal mucosal cells. Similarly, specific antibodies can bind to certain toxins, blocking them from attaching to target cells and thus neutralizing their toxic effects. Viruses can be neutralized and prevented from infecting a cell by the same mechanism (figure 1.25).
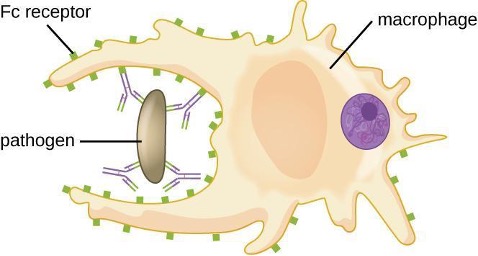
As described in section 1.4, opsonization is the coating of a pathogen with molecules, such as complement factors, C-reactive protein, and serum amyloid A, to assist in phagocyte binding to facilitate phagocytosis. IgG antibodies also serve as excellent opsonins, binding their Fab sites to specific epitopes on the surface of pathogens. Phagocytic cells such as macrophages, dendritic cells, and neutrophils have receptors on their surfaces that recognize and bind to the Fc portion of the IgG molecules; thus, IgG helps such phagocytes attach to and engulf the pathogens they have bound (figure 1.26).
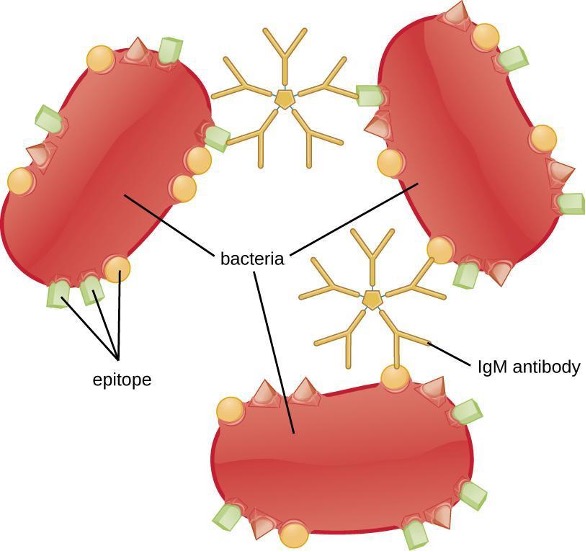
Agglutination or aggregation involves the cross-linking of pathogens by antibodies to create large aggregates (figure 1.27). IgG has two Fab antigen-binding sites, which can bind to two separate pathogen cells, clumping them together. When multiple IgG antibodies are involved, large aggregates can develop; these aggregates are easier for the kidneys and spleen to filter from the blood and easier for phagocytes to ingest for destruction. The pentameric structure of IgM provides ten Fab binding sites per molecule, making it the most efficient antibody for agglutination.
Another important function of antibodies is activation of the complement cascade. The complement system is an important component of the innate defenses, promoting the inflammatory response, recruiting phagocytes to site of infection, enhancing phagocytosis by opsonization, and killing gram-negative bacterial pathogens with the membrane attack complex (MAC). Complement activation can occur through three different pathways (see figure 1.7), but the most efficient is the classical pathway, which requires the initial binding of IgG or IgM antibodies to the surface of a pathogen cell, allowing for recruitment and activation of the C1 complex.
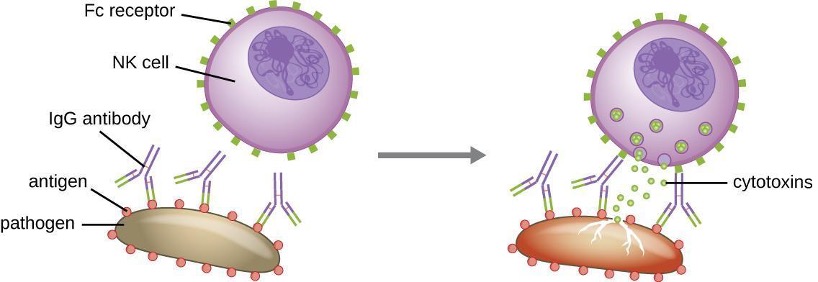
Yet another important function of antibodies is antibody-dependent cell-mediated cytotoxicity (ADCC), which enhances killing of pathogens that are too large to be phagocytosed. This process is best characterized for natural killer cells (NK cells), as shown in figure 1.28, but it can also involve macrophages and eosinophils. ADCC occurs when the Fab region of an IgG antibody binds to a large pathogen; Fc receptors on effector cells (e.g., NK cells) then bind to the Fc region of the antibody, bringing them into close proximity with the target pathogen. The effector cell then secretes powerful cytotoxins (e.g., perforin and granzymes) that kill the pathogen.
1.9 Major Histocompatibility Complexes and Antigen-Presenting Cells
As discussed in section 1.5, major histocompatibility complex (MHC) molecules are expressed on the surface of healthy cells, identifying them as normal and “self” to natural killer (NK) cells. MHC molecules also play an important role in the presentation of foreign antigens, which is a critical step in the activation of T cells and thus an important mechanism of the adaptive immune system.
Major Histocompatibility Complex Molecules
The major histocompatibility complex (MHC) is a collection of genes coding for MHC molecules found on the surface of all nucleated cells of the body. In humans, the MHC genes are also referred to as human leukocyte antigen (HLA) genes. Mature red blood cells, which lack a nucleus, are the only cells that do not express MHC molecules on their surface.
There are two classes of MHC molecules involved in adaptive immunity, MHC I and MHC II (figure 1.29). MHC I molecules are found on all nucleated cells; they present normal self-antigens as well as abnormal or nonself pathogens to the effector T cells involved in cellular immunity. In contrast, MHC II molecules are only found on macrophages, dendritic cells, and B cells; they present abnormal or nonself pathogen antigens for the initial activation of T cells.
Both types of MHC molecules are transmembrane glycoproteins that assemble as dimers in the cytoplasmic membrane of cells, but their structures are quite different. MHC I molecules are composed of a longer α protein chain coupled with a smaller β2 microglobulin protein, and only the α chain spans the cytoplasmic membrane. The α chain of the MHC I molecule folds into three separate domains: α1, α2 and α3. MHC II molecules are composed of two protein chains (an α and a β chain) that are approximately similar in length. Both chains of the MHC II molecule possess portions that span the plasma membrane, and each chain folds into two separate domains: α1 and α2, and β1, and β2. In order to present abnormal or non-self-antigens to T cells, MHC molecules have a cleft that serves as the antigen-binding site near the top (or outermost) portion of the MHC I or MHC II dimer. For MHC I, the antigen-binding cleft is formed by the α1 and α2 domains, whereas for MHC II, the cleft is formed by the α1 and β1 domains (figure 1.32).
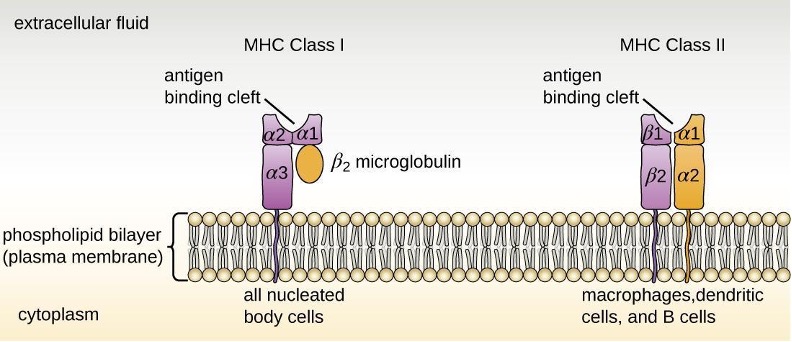
Antigen-Presenting Cells (APCs)
All nucleated cells in the body have mechanisms for processing and presenting antigens in association with MHC molecules. This signals the immune system, indicating whether the cell is normal and healthy or infected with an intracellular pathogen. However, only macrophages, dendritic cells, and B cells have the ability to present antigens specifically for the purpose of activating T cells; for this reason, these types of cells are sometimes referred to as antigen-presenting cells (APCs).
While all APCs play a similar role in adaptive immunity, there are some important differences to consider. Macrophages and dendritic cells are phagocytes that ingest and kill pathogens that penetrate the first-line barriers (i.e., skin and mucous membranes). B cells, on the other hand, do not function as phagocytes but play a primary role in the production and secretion of antibodies. In addition, whereas macrophages and dendritic cells recognize pathogens through nonspecific receptor interactions (e.g., PAMPs, toll-like receptors, and receptors for opsonizing complement or antibody), B cells interact with foreign pathogens or their free antigens using antigen-specific immunoglobulin as receptors (monomeric IgD and IgM). When the immunoglobulin receptors bind to an antigen, the B cell internalizes the antigen by endocytosis before processing and presenting the antigen to T cells.
Antigen Presentation with MHC II Molecules
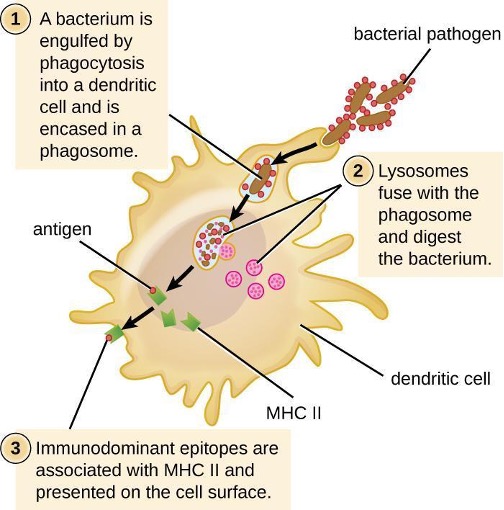
MHC II molecules are only found on the surface of APCs. Macrophages and dendritic cells use similar mechanisms for processing and presentation of antigens and their epitopes in association with MHC II; B cells use somewhat different mechanisms that will be described further in section 1.11. For now, we will focus on the steps of the process as they pertain to dendritic cells.
After a dendritic cell recognizes and attaches to a pathogen cell, the pathogen is internalized by phagocytosis and is initially contained within a phagosome. Lysosomes containing antimicrobial enzymes and chemicals fuse with the phagosome to create a phagolysosome, where degradation of the pathogen for antigen processing begins. Proteases (protein-degrading) are especially important in antigen processing because only protein antigen epitopes are presented to T cells by MHC II (figure 1.30).
APCs do not present all possible epitopes to T cells; only a selection of the most antigenic or immunodominant epitopes are presented. The mechanism by which epitopes are selected for processing and presentation by an APC is complicated and not well understood; however, once the most antigenic, immunodominant epitopes have been processed, they associate within the antigen-binding cleft of MHC II molecules and are translocated to the cell surface of the dendritic cell for presentation to T cells.
Antigen Presentation with MHC I Molecules
MHC I molecules, found on all normal, healthy, nucleated cells, signal to the immune system that the cell is a normal “self” cell. In a healthy cell, proteins normally found in the cytoplasm are degraded by proteasomes (enzyme complexes responsible for degradation and processing of proteins) and processed into self-antigen epitopes; these self-antigen epitopes bind within the MHC I antigen-binding cleft and are then presented on the cell surface. Immune cells, such as NK cells, recognize these self-antigens and do not target the cell for destruction. However, if a cell becomes infected with an intracellular pathogen (e.g., a virus), protein antigens specific to the pathogen are processed in the proteasomes and bind with MHC I molecules for presentation on the cell surface. This presentation of pathogen-specific antigens with MHC I signals that the infected cell must be targeted for destruction along with the pathogen.
Before elimination of infected cells can begin, APCs must first activate the T cells involved in cellular immunity. If an intracellular pathogen directly infects the cytoplasm of an APC, then the processing and presentation of antigens can occur as described (in proteasomes and on the cell surface with MHC I). However, if the intracellular pathogen does not directly infect APCs, an alternative strategy called cross-presentation is utilized. In cross-presentation, antigens are brought into the APC by mechanisms normally leading to presentation with MHC II (i.e., through phagocytosis), but the antigen is presented on an MHC I molecule for CD8 T cells. The exact mechanisms by which cross-presentation occurs are not yet well understood, but it appears that cross-presentation is primarily a function of dendritic cells and not macrophages or B cells.
1.10 T Lymphocytes and Cellular Immunity

As explained in section 1.8, the antibodies involved in humoral immunity often bind pathogens and toxins before they can attach to and invade host cells. Thus, humoral immunity is primarily concerned with fighting pathogens in extracellular spaces. However, pathogens that have already gained entry to host cells are largely protected from the humoral antibody-mediated defenses. Cellular immunity, on the other hand, targets and eliminates intracellular pathogens through the actions of T lymphocytes, or T cells (figure 1.31). T cells also play a more central role in orchestrating the overall adaptive immune response (humoral as well as cellular) along with the cellular defenses of innate immunity.
T Cell Production and Maturation
T cells, like all other white blood cells involved in innate and adaptive immunity, are formed from multipotent hematopoietic stem cells (HSCs) in the bone marrow (see figure 1.9). However, unlike the white blood cells of innate immunity, eventual T cells differentiate first into lymphoid stem cells that then become small, immature lymphocytes, sometimes called lymphoblasts. The first steps of differentiation occur in the red marrow of bones (figure 1.32), after which immature T lymphocytes enter the bloodstream and travel to the thymus for the final steps of maturation (figure 1.33). Once in the thymus, the immature T lymphocytes are referred to as thymocytes.
The maturation of thymocytes within the thymus can be divided into three critical steps of positive and negative selection, collectively referred to as thymic selection. The first step of thymic selection occurs in the cortex of the thymus and involves the development of a functional T-cell receptor (TCR) that is required for activation by APCs. Thymocytes with defective TCRs are removed by negative selection through the induction of apoptosis (programmed controlled cell death). The second step of thymic selection also occurs in the cortex and involves the positive selection of thymocytes that will interact appropriately with MHC molecules. Thymocytes that can interact appropriately with MHC molecules receive a positive stimulation that moves them further through the process of maturation, whereas thymocytes that do not interact appropriately are not stimulated and are eliminated by apoptosis. The third and final step of thymic selection occurs in both the cortex and medulla and involves negative selection to remove self-reacting thymocytes, those that react to self-antigens, by apoptosis. This final step is sometimes referred to as central tolerance because it prevents self-reacting T cells from reaching the bloodstream and potentially causing autoimmune disease, which occurs when the immune system attacks healthy “self” cells.
Despite central tolerance, some self-reactive T cells generally escape the thymus and enter the peripheral bloodstream. Therefore, a second line of defense called peripheral tolerance is needed to protect against autoimmune disease. Peripheral tolerance involves mechanisms of anergy and inhibition of self-reactive T cells by regulatory T cells. Anergy refers to a state of nonresponsiveness to antigen stimulation. In the case of self-reactive T cells that escape the thymus, lack of an essential co-stimulatory signal required for activation causes anergy and prevents autoimmune activation. Regulatory T cells participate in peripheral tolerance by inhibiting the activation and function of self-reactive T cells and by secreting anti-inflammatory cytokines.
It is not completely understood what events specifically direct maturation of thymocytes into regulatory T cells. Current theories suggest the critical events may occur during the third step of thymic selection, when most self-reactive T cells are eliminated. Regulatory T cells may receive a unique signal that is below the threshold required to target them for negative selection and apoptosis. Consequently, these cells continue to mature and then exit the thymus, armed to inhibit the activation of self-reactive T cells.
It has been estimated that the three steps of thymic selection eliminate 98% of thymocytes. The remaining 2% that exit the thymus migrate through the bloodstream and lymphatic system to sites of secondary lymphoid organs/tissues, such as the lymph nodes, spleen, and tonsils (figure 1.33), where they await activation through the presentation of specific antigens by APCs. Until they are activated, they are known as mature naïve T cells.
Classes of T Cells
T cells can be categorized into three distinct classes: helper T cells, regulatory T cells, and cytotoxic T cells. These classes are differentiated based on their expression of certain surface molecules, their mode of activation, and their functional roles in adaptive immunity (table 1.10).
All T cells produce a cluster of differentiation (CD) molecules, cell surface glycoproteins that can be used to identify and distinguish between the various types of white blood cells. Although T cells can produce a variety of CD molecules, CD4 and CD8 are the two most important used for differentiation of the classes. Helper T cells and regulatory T cells are characterized by the expression of CD4 on their surface, whereas cytotoxic T cells are characterized by the expression of CD8.
Classes of T cells can also be distinguished by the specific MHC molecules and APCs with which they interact for activation. Helper T cells and regulatory T cells can only be activated by APCs presenting antigens associated with MHC II. In contrast, cytotoxic T cells recognize antigens presented in association with MHC I, either by APCs or by nucleated cells infected with an intracellular pathogen.
The different classes of T cells also play different functional roles in the immune system. Helper T cells serve as the central orchestrators that help activate and direct functions of humoral and cellular immunity. In addition, helper T cells enhance the pathogen-killing functions of macrophages and NK cells of innate immunity. In contrast, the primary role of regulatory T cells is to prevent undesirable and potentially damaging immune responses. Their role in peripheral tolerance, for example, protects against autoimmune disorders, as discussed earlier. Finally, cytotoxic T cells are the primary effector cells for cellular immunity. They recognize and target cells that have been infected by intracellular pathogens, destroying infected cells along with the pathogens inside.
| Class | Surface CD Molecules | Activation | Functions |
|---|---|---|---|
| Helper T cells | CD4 | APCs presenting antigens associated with MHC II | Orchestrate humoral and cellular immunity |
| Involved in the activation of macrophages and NK cells | |||
| Regulatory T cells | CD4 | APCs presenting antigens associated with MHC II | Involved in peripheral tolerance and prevention of autoimmune responses |
| Cytotoxic T cells | CD8 | APCs or infected nucleated cells presenting antigens associated with MHC I | Destroy cells infected with intracellular pathogens |
Table 1.10: Classes of T cells
T-Cell Receptors
For both helper T cells and cytotoxic T cells, activation is a complex process that requires the interactions of multiple molecules and exposure to cytokines. The T-cell receptor (TCR) is involved in the first step of pathogen epitope recognition during the activation process.
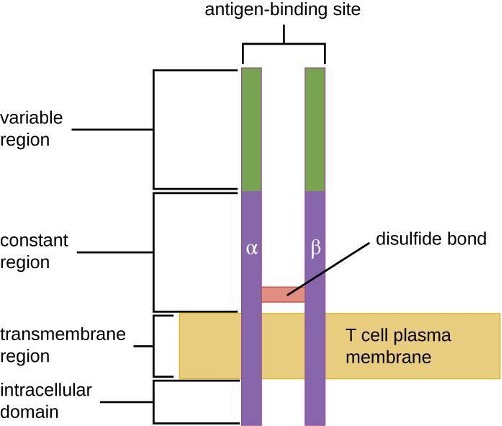
The TCR comes from the same receptor family as the antibodies IgD and IgM, the antigen receptors on the B cell membrane surface, and thus shares common structural elements. Similar to antibodies, the TCR has a variable region and a constant region, and the variable region provides the antigen-binding site (figure 1.34). However, the structure of TCR is smaller and less complex than the immunoglobulin molecules (figure 1.24). Whereas immunoglobulins have four peptide chains and Y-shaped structures, the TCR consists of just two peptide chains (α and β chains), both of which span the cytoplasmic membrane of the T cell.
TCRs are epitope-specific, and it has been estimated that 25 million T cells with unique epitope-binding TCRs are required to protect an individual against a wide range of microbial pathogens. Because the human genome only contains about 25,000 genes, we know that each specific TCR cannot be encoded by its own set of genes. This raises the question of how such a vast population of T cells with millions of specific TCRs can be achieved. The answer is a process called genetic rearrangement, which occurs in the thymus during the first step of thymic selection.
The genes that code for the variable regions of the TCR are divided into distinct gene segments called variable (V), diversity (D), and joining (J) segments. The genes segments associated with the α chain of the TCR consist of 70 or more different Vα segments and 61 different Jα segments. The gene segments associated with the β chain of the TCR consist of 52 different Vβ segments, two different Dβ segments, and 13 different Jβ segments. During the development of the functional TCR in the thymus, genetic rearrangement in a T cell brings together one Vα segment and one Jα segment to code for the variable region of the α chain. Similarly, genetic rearrangement brings one of the Vβ segments together with one of the Dβ segments and one of the Jβ segments to code for the variable region of the β chain. All the possible combinations of rearrangements between different segments of V, D, and J provide the genetic diversity required to produce millions of TCRs with unique epitope-specific variable regions.
Activation and Differentiation of Helper T Cells
Helper T cells can only be activated by APCs presenting processed foreign epitopes in association with MHC II. The first step in the activation process is TCR recognition of the specific foreign epitope presented within the MHC II antigen-binding cleft. The second step involves the interaction of CD4 on the helper T cell with a region of the MHC II molecule separate from the antigen-binding cleft. This second interaction anchors the MHC II-TCR complex and ensures that the helper T cell is recognizing both the foreign (“nonself”) epitope and “self” antigen of the APC; both recognitions are required for activation of the cell. In the third step, the APC and T cell secrete cytokines that activate the helper T cell. The activated helper T cell then proliferates, dividing by mitosis to produce clonal naïve helper T cells that differentiate into subtypes with different functions (figure 1.35).
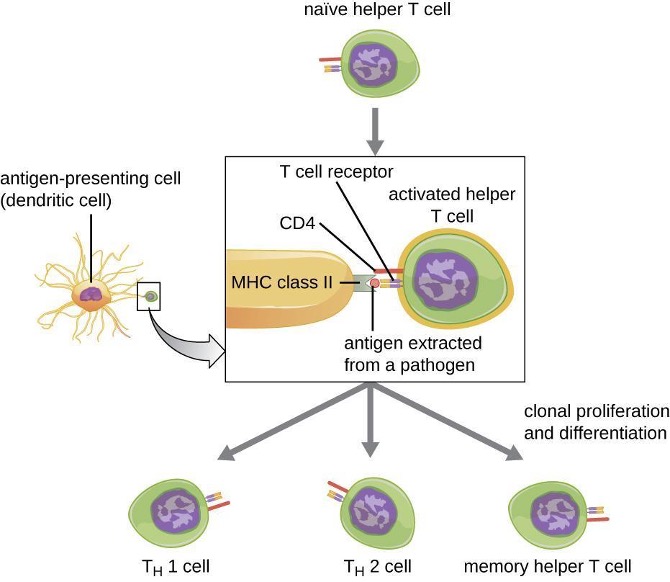
Activated helper T cells can differentiate into one of four distinct subtypes, summarized in table 1.11. The differentiation process is directed by APC-secreted cytokines. Depending on which APC-secreted cytokines interact with an activated helper T cell, the cell may differentiate into a T helper 1 (TH1) cell, a T helper 2 (TH2) cell, or a memory helper T cell. The two types of helper T cells are relatively short-lived effector cells, meaning that they perform various functions of the immediate immune response. In contrast, memory helper T cells are relatively long lived; they are programmed to “remember” a specific antigen or epitope in order to mount a rapid, strong, secondary response to subsequent exposures.
TH1 cells secrete their own cytokines that are involved in stimulating and orchestrating other cells involved in adaptive and innate immunity. For example, they stimulate cytotoxic T cells, enhancing their killing of infected cells and promoting differentiation into memory cytotoxic T cells. TH1 cells also stimulate macrophages and neutrophils to become more effective in their killing of intracellular bacteria. They can also stimulate NK cells to become more effective at killing target cells.
TH2 cells play an important role in orchestrating the humoral immune response through their secretion of cytokines that activate B cells and direct B cell differentiation and antibody production. Various cytokines produced by TH2 cells orchestrate antibody class switching, which allows B cells to switch between the production of IgM, IgG, IgA, and IgE as needed to carry out specific antibody functions and to provide pathogen-specific humoral immune responses.
A third subtype of helper T cells called TH17 cells was discovered through observations that immunity to some infections is not associated with TH1 or TH2 cells. TH17 cells and the cytokines they produce appear to be specifically responsible for the body’s defense against chronic mucocutaneous infections. Patients who lack sufficient TH17 cells in the mucosa (e.g., HIV patients) may be more susceptible to bacteremia and gastrointestinal infections.[2]
| Subtype | Functions |
|---|---|
| TH1 cells | Stimulate cytotoxic T cells and produce memory cytotoxic T cells |
| Stimulate macrophages and neutrophils (PMNs) for more effective intracellular killing of pathogens | |
| Stimulate NK cells to kill more effectively | |
| TH2 cells | Stimulate B cell activation and differentiation into plasma cells and memory B cells |
| Direct antibody class switching in B cells | |
| TH17 cells | Stimulate immunity to specific infections such as chronic mucocutaneous infections |
| Memory helper T cells | “Remember” a specific pathogen and mount a strong, rapid secondary response upon re-exposure |
Table 1.11: Subtypes of helper T cells
Activation and Differentiation of Cytotoxic T Cells
Cytotoxic T cells (also referred to as cytotoxic T lymphocytes, or CTLs) are activated by APCs in a three-step process similar to that of helper T cells. The key difference is that the activation of cytotoxic T cells involves recognition of an antigen presented with MHC I (as opposed to MHC II) and interaction of CD8 (as opposed to CD4) with the receptor complex. After the successful co-recognition of foreign epitope and self-antigen, the production of cytokines by the APC and the cytotoxic T cell activates clonal proliferation and differentiation. Activated cytotoxic T cells can differentiate into effector cytotoxic T cells that target pathogens for destruction or memory cells that are ready to respond to subsequent exposures.
As noted, proliferation and differentiation of cytotoxic T cells is also stimulated by cytokines secreted from TH1 cells activated by the same foreign epitope. The co-stimulation that comes from these TH1 cells is provided by secreted cytokines. Although it is possible for activation of cytotoxic T cells to occur without stimulation from TH1 cells, the activation is not as effective or long-lasting.
Once activated, cytotoxic T cells serve as the effector cells of cellular immunity, recognizing and killing cells infected with intracellular pathogens through a mechanism very similar to that of NK cells. However, whereas NK cells recognize nonspecific signals of cell stress or abnormality, cytotoxic T cells recognize infected cells through antigen presentation of pathogen-specific epitopes associated with MHC I. Once an infected cell is recognized, the TCR of the cytotoxic T cell binds to the epitope and releases perforin and granzymes that destroy the infected cell (figure 1.36). Perforin is a protein that creates pores in the target cell, and granzymes are proteases that enter the pores and induce apoptosis. This mechanism of programmed cell death is a controlled and efficient means of destroying and removing infected cells without releasing the pathogens inside to infect neighboring cells, as might occur if the infected cells were simply lysed.
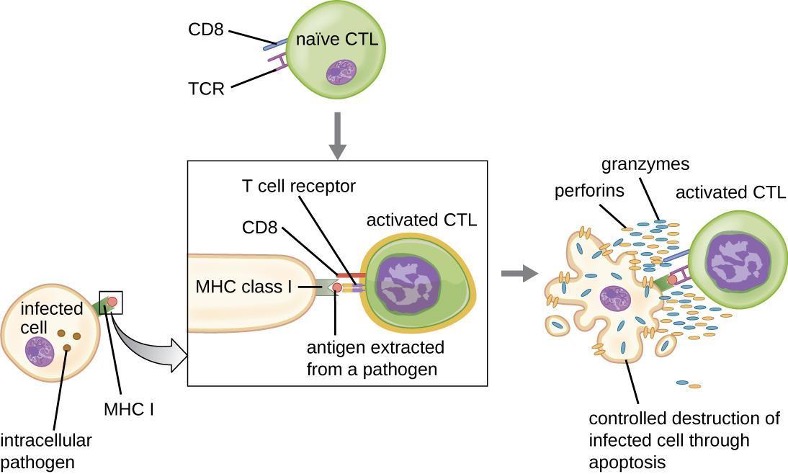
Superantigens and Unregulated Activation of T Cells
When T cell activation is controlled and regulated, the result is a protective response that is effective in combating infections. However, if T cell activation is unregulated and excessive, the result can be life-threatening. Certain bacterial and viral pathogens produce toxins known as superantigens (see section 2.14) that can trigger such an unregulated response. Known bacterial superantigens include toxic shock syndrome toxin (TSST), staphylococcal enterotoxins, streptococcal pyrogenic toxins, streptococcal superantigen, and the streptococcal mitogenic exotoxin. Viruses known to produce superantigens include Epstein-Barr virus (human herpesvirus 4), cytomegalovirus (human herpesvirus 5), among others.
The mechanism of T cell activation by superantigens involves their simultaneous binding to MHC II molecules of APCs and the variable region of the TCR β chain. This binding occurs outside of the antigen-binding cleft of MHC II, so the superantigen will bridge together and activate MHC II and TCR without specific foreign epitope recognition (figure 1.37). The result is an excessive, uncontrolled release of cytokines, often called a cytokine storm, which stimulates an excessive inflammatory response. This can lead to a dangerous decrease in blood pressure, shock, multi-organ failure, and potentially, death.
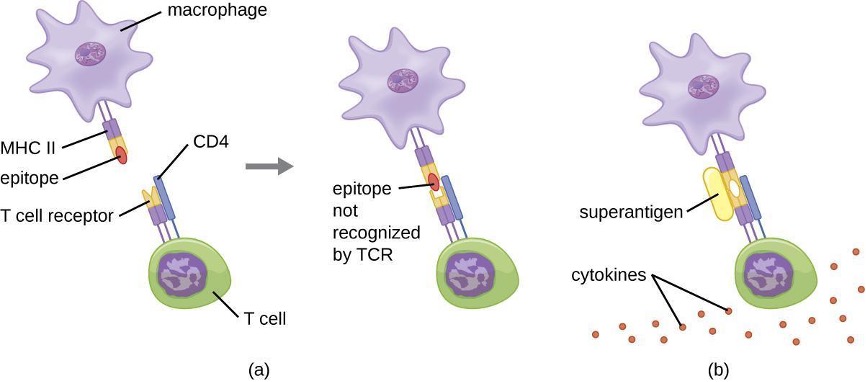
1.11 B Lymphocytes and Humoral Immunity
Humoral immunity refers to mechanisms of the adaptive immune defenses that are mediated by antibodies secreted by B lymphocytes, or B cells. This section will focus on B cells and discuss their production and maturation, receptors, and mechanisms of activation.
B Cell Production and Maturation
Like T cells, B cells are formed from multipotent hematopoietic stem cells (HSCs) in the bone marrow and follow a pathway through lymphoid stem cells and lymphoblast (see figure 1.9). Unlike T cells, however, lymphoblasts destined to become B cells do not leave the bone marrow and travel to the thymus for maturation. Rather, eventual B cells continue to mature in the bone marrow.
The first step of B cell maturation is an assessment of the functionality of their antigen-binding receptors. This occurs through positive selection for B cells with normal functional receptors. A mechanism of negative selection is then used to eliminate self-reacting B cells and minimize the risk of autoimmunity. Negative selection of self-reacting B cells can involve elimination by apoptosis, editing or modification of the receptors so they are no longer self-reactive, or induction of anergy in the B cell. Immature B cells that pass the selection in the bone marrow then travel to the spleen for their final stages of maturation. There they become naïve mature B cells, i.e., mature B cells that have not yet been activated.
B-Cell Receptors
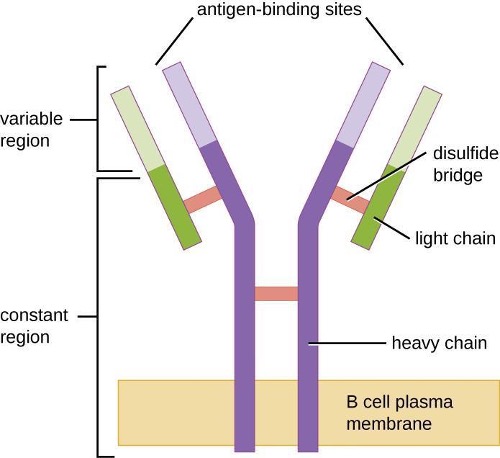
Like T cells, B cells possess antigen-specific receptors with diverse specificities. Although they rely on T cells for optimum function, B cells can be activated without help from T cells. B-cell receptors (BCRs) for naïve mature B cells are membrane-bound monomeric forms of IgD and IgM. They have two identical heavy chains and two identical light chains connected by disulfide bonds into a basic Y shape (figure 1.38). The trunk of the Y-shaped molecule, the constant region of the two heavy chains, spans the B cell membrane. The two antigen-binding sites exposed to the exterior of the B cell are involved in the binding of specific pathogen epitopes to initiate the activation process. It is estimated that each naïve mature B cell has upwards of 100,000 BCRs on its membrane, and each of these BCRs has an identical epitope-binding specificity.
In order to be prepared to react to a wide range of microbial epitopes, B cells, like T cells, use genetic rearrangement of hundreds of gene segments to provide the necessary diversity of receptor specificities. The variable region of the BCR heavy chain is made up of V, D, and J segments, similar to the β chain of the TCR. The variable region of the BCR light chain is made up of V and J segments, similar to the α chain of the TCR. Genetic rearrangement of all possible combinations of V-J-D (heavy chain) and V-J (light chain) provides for millions of unique antigen-binding sites for the BCR and for the antibodies secreted after activation.
One important difference between BCRs and TCRs is the way they can interact with antigenic epitopes. Whereas TCRs can only interact with antigenic epitopes that are presented within the antigen-binding cleft of MHC I or MHC II, BCRs do not require antigen presentation with MHC; they can interact with epitopes on free antigens or with epitopes displayed on the surface of intact pathogens. Another important difference is that TCRs only recognize protein epitopes, whereas BCRs can recognize epitopes associated with different molecular classes (e.g., proteins, polysaccharides, lipopolysaccharides).
Activation of B cells occurs through different mechanisms depending on the molecular class of the antigen. Activation of a B cell by a protein antigen requires the B cell to function as an APC, presenting the protein epitopes with MHC II to helper T cells. Because of their dependence on T cells for activation of B cells, protein antigens are classified as T-dependent antigens. In contrast, polysaccharides, lipopolysaccharides, and other nonprotein antigens are considered T-independent antigens because they can activate B cells without antigen processing and presentation to T cells.
T Cell-Independent Activation of B cells
Activation of B cells without the cooperation of helper T cells is referred to as T cell-independent activation and occurs when BCRs interact with T-independent antigens. T-independent antigens (e.g., polysaccharide capsules, lipopolysaccharide) have repetitive epitope units within their structure, and this repetition allows for the cross-linkage of multiple BCRs, providing the first signal for activation (figure 1.39). Because T cells are not involved, the second signal has to come from other sources, such as interactions of toll-like receptors with PAMPs or interactions with factors from the complement system.
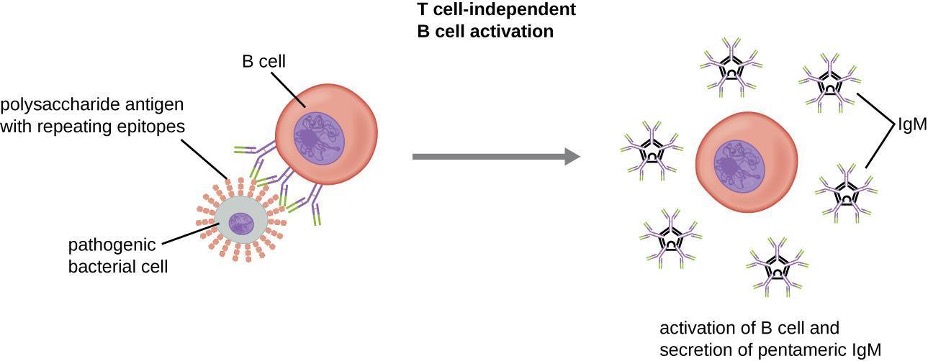
Once a B cell is activated, it undergoes clonal proliferation and daughter cells differentiate into plasma cells. Plasma cells are antibody factories that secrete large quantities of antibodies. After differentiation, the surface BCRs disappear and the plasma cell secretes pentameric IgM molecules that have the same antigen specificity as the BCRs (figure 1.39).
The T cell-independent response is short-lived and does not result in the production of memory B cells. Thus it will not result in a secondary response to subsequent exposures to T-independent antigens.
T Cell-Dependent Activation of B cells
T cell-dependent activation of B cells is more complex than T cell-independent activation, but the resulting immune response is stronger and develops memory. T cell-dependent activation can occur either in response to free protein antigens or to protein antigens associated with an intact pathogen. Interaction between the BCRs on a naïve mature B cell and a free protein antigen stimulates internalization of the antigen, whereas interaction with antigens associated with an intact pathogen initiates the extraction of the antigen from the pathogen before internalization. Once internalized inside the B cell, the protein antigen is processed and presented with MHC II. The presented antigen is then recognized by helper T cells specific to the same antigen. The TCR of the helper T cell recognizes the foreign antigen, and the T cell’s CD4 molecule interacts with MHC II on the B cell. The coordination between B cells and helper T cells that are specific to the same antigen is referred to as linked recognition.
Once activated by linked recognition, TH2 cells produce and secrete cytokines that activate the B cell and cause proliferation into clonal daughter cells. After several rounds of proliferation, additional cytokines provided by the TH2 cells stimulate the differentiation of activated B cell clones into memory B cells, which will quickly respond to subsequent exposures to the same protein epitope, and plasma cells that lose their membrane BCRs and initially secrete pentameric IgM (figure 1.40).
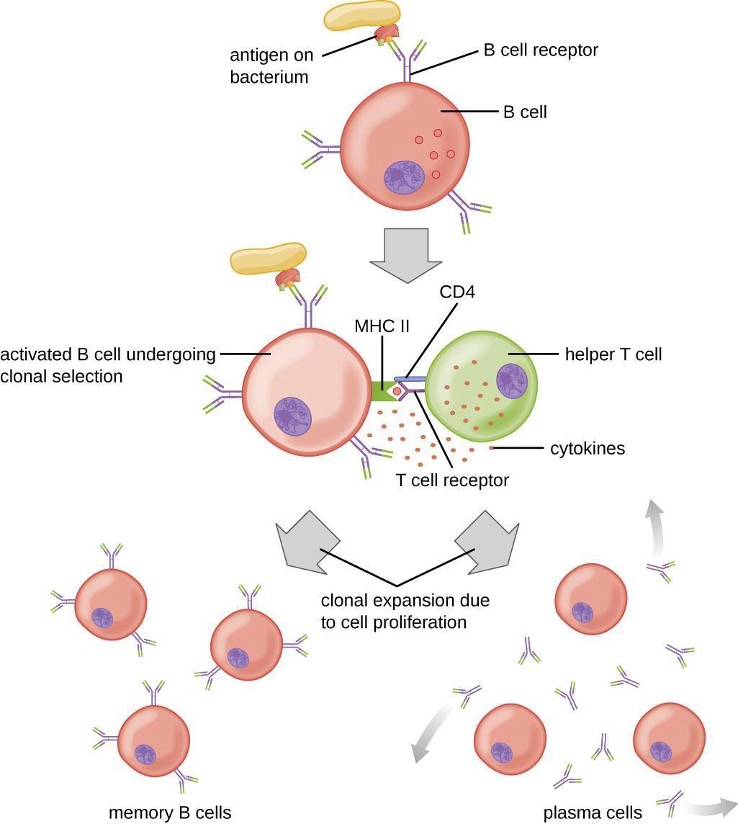
After initial secretion of IgM, cytokines secreted by TH2 cells stimulate the plasma cells to switch from IgM production to production of IgG, IgA, or IgE. This process, called class switching or isotype switching, allows plasma cells cloned from the same activated B cell to produce a variety of antibody classes with the same epitope specificity. Class switching is accomplished by genetic rearrangement of gene segments encoding the constant region, which determines an antibody’s class. The variable region is not changed, so the new class of antibody retains the original epitope specificity.
Primary and Secondary Responses
T cell-dependent activation of B cells plays an important role in both the primary and secondary responses associated with adaptive immunity. With the first exposure to a protein antigen, a T cell-dependent primary antibody response occurs. The initial stage of the primary response is a lag period, or latent period, of approximately 10 days, during which no antibody can be detected in serum. This lag period is the time required for all of the steps of the primary response, including naïve mature B cells’ antigens binding with BCRs, antigen processing and presentation, helper T cell activation, B cell activation, and clonal proliferation. The end of the lag period is characterized by a rise in IgM levels in the serum, as TH2 cells stimulate B cell differentiation into plasma cells. IgM levels reach their peak around 14 days after primary antigen exposure; at about this same time, TH2 stimulates antibody class switching, and IgM levels in serum begin to decline. Meanwhile, levels of IgG increase until they reach a peak about three weeks into the primary response (figure 1.41).
During the primary response, some of the cloned B cells are differentiated into memory B cells programmed to respond to subsequent exposures. This secondary response occurs more quickly and forcefully than the primary response. The lag period is decreased to only a few days and the production of IgG is significantly higher than observed for the primary response (figure 1.41). In addition, the antibodies produced during the secondary response are more effective and bind with higher affinity to the targeted epitopes. Plasma cells produced during secondary responses live longer than those produced during the primary response, so levels of specific antibodies remain elevated for a longer period of time.
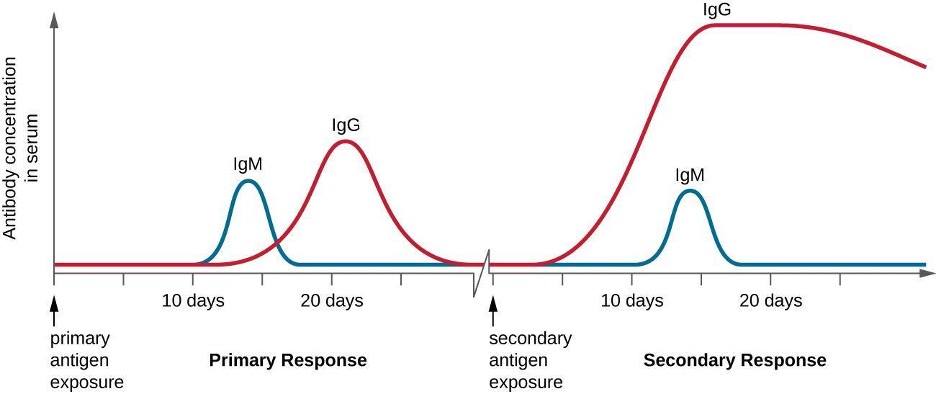
1.12 Vaccines
For many diseases, prevention is the best form of treatment, and few strategies for disease prevention are as effective as vaccination. Vaccination is a form of artificial immunity. By artificially stimulating the adaptive immune defenses, a vaccine triggers memory cell production similar to that which would occur during a primary response. In so doing, the patient is able to mount a strong secondary response upon exposure to the pathogen—but without having to first suffer through an initial infection. In this section, we will explore several different kinds of artificial immunity along with various types of vaccines and the mechanisms by which they induce artificial immunity.
Classifications of Adaptive Immunity
All forms of adaptive immunity can be described as either active or passive. Active immunity refers to the activation of an individual’s own adaptive immune defenses, whereas passive immunity refers to the transfer of adaptive immune defenses from another individual or animal. Active and passive immunity can be further subdivided based on whether the protection is acquired naturally or artificially.
Natural active immunity is adaptive immunity that develops after natural exposure to a pathogen (table 1.12). Examples would include the lifelong immunity that develops after recovery from a chickenpox or measles infection (although an acute infection is not always necessary to activate adaptive immunity). The length of time that an individual is protected can vary substantially depending upon the pathogen and antigens involved. For example, activation of adaptive immunity by protein spike structures during an intracellular viral infection can activate lifelong immunity, whereas activation by carbohydrate capsule antigens during an extracellular bacterial infection may activate shorter-term immunity.
| Natural acquired | Artificial required | |
|---|---|---|
| Passive | Immunity acquired from antibodies passed in breast milk or through placenta |
Immunity gained through antibodies harvested from another person or an animal |
| Active | Immunity gained through illness and recovery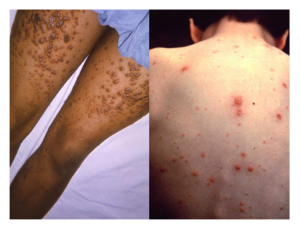 |
Immunity acquired through a vaccine |
Table 1.12: The four mechanisms of acquisition of immunity.
Natural passive immunity involves the natural passage of antibodies from a mother to her child before and after birth. IgG is the only antibody class that can cross the placenta from mother’s blood to the fetal blood supply. Placental transfer of IgG is an important passive immune defense for the infant, lasting up to six months after birth. Secretory IgA can also be transferred from mother to infant through breast milk.
Artificial passive immunity refers to the transfer of antibodies produced by a donor (human or animal) to another individual. This transfer of antibodies may be done as a prophylactic measure (i.e., to prevent disease after exposure to a pathogen) or as a strategy for treating an active infection. For example, artificial passive immunity is commonly used for post-exposure prophylaxis against rabies, hepatitis A, hepatitis B, and chickenpox (in high-risk individuals). Active infections treated by artificial passive immunity include cytomegalovirus infections in immunocompromised patients and Ebola virus infections. In 1995, eight patients in the Democratic Republic of the Congo with active Ebola infections were treated with blood transfusions from patients who were recovering from Ebola. Only one of the eight patients died (a 12.5% mortality rate), which was much lower than the expected 80% mortality rate for Ebola in untreated patients.[3]Artificial passive immunity is also used for the treatment of diseases caused by bacterial toxins, including tetanus, botulism, and diphtheria.
Artificial active immunity is the foundation for vaccination. It involves the activation of adaptive immunity through the deliberate exposure of an individual to weakened or inactivated pathogens, or preparations consisting of key pathogen antigens.
Herd Immunity
The four kinds of immunity just described result from an individual’s adaptive immune system. For any given disease, an individual may be considered immune or susceptible depending on his or her ability to mount an effective immune response upon exposure. Thus, any given population is likely to have some individuals who are immune and other individuals who are susceptible. If a population has very few susceptible individuals, even those susceptible individuals will be protected by a phenomenon called herd immunity. Herd immunity has nothing to do with an individual’s ability to mount an effective immune response; rather, it occurs because there are too few susceptible individuals in a population for the disease to spread effectively.
Vaccination programs create herd immunity by greatly reducing the number of susceptible individuals in a population. Even if some individuals in the population are not vaccinated, as long as a certain percentage is immune (either naturally or artificially), the few susceptible individuals are unlikely to be exposed to the pathogen. However, because new individuals are constantly entering populations (for example, through birth or relocation), vaccination programs are necessary to maintain herd immunity.
Variolation and Vaccination
Thousands of years ago, it was first recognized that individuals who survived a smallpox infection were immune to subsequent infections. Variolation refers to the deliberate inoculation of individuals with infectious material from scabs or pustules of smallpox victims. Infectious materials were either injected into the skin or introduced through the nasal route. The infection that developed was usually milder than naturally acquired smallpox, and recovery from the milder infection provided protection against the more serious disease.
Although the majority of individuals treated by variolation developed only mild infections, the practice was not without risks. More serious and sometimes fatal infections did occur, and because smallpox was contagious, infections resulting from variolation could lead to epidemics. Even so, the practice of variolation for smallpox prevention spread to other regions, including India, Africa, and Europe.
Although variolation had been practiced for centuries, the English physician Edward Jenner (1749–1823) is generally credited with developing the modern process of vaccination. Jenner observed that milkmaids who developed cowpox, a disease similar to smallpox but milder, were immune to the more serious smallpox. This led Jenner to hypothesize that exposure to a less virulent pathogen could provide immune protection against a more virulent pathogen, providing a safer alternative to variolation. In 1796, Jenner tested his hypothesis by obtaining infectious samples from a milkmaid’s active cowpox lesion and injecting the materials into a young boy (figure 1.42). The boy developed a mild infection that included a low-grade fever, discomfort in his axillae (armpit), and loss of appetite. When the boy was later exposed to infectious samples from smallpox lesions, he did not contract smallpox.[4] This new approach was termed vaccination, a name deriving from the use of cowpox (Latin vacca meaning “cow”) to protect against smallpox. Today, we know that Jenner’s vaccine worked because the cowpox virus is genetically and antigenically related to the Variola viruses that caused smallpox. Exposure to cowpox antigens resulted in a primary response and the production of memory cells that had identical or related epitopes of Variola virus upon a later exposure to smallpox.
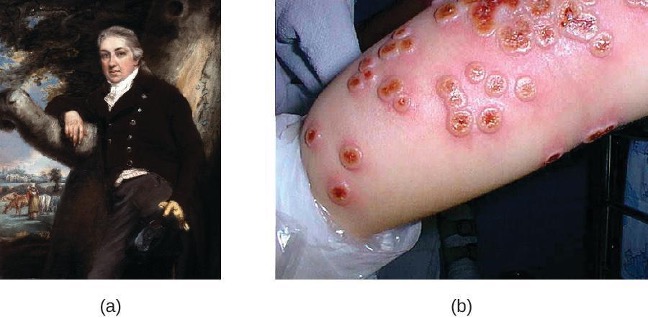
The success of Jenner’s smallpox vaccination led other scientists to develop vaccines for other diseases. Perhaps the most notable was Louis Pasteur, who developed vaccines for rabies, cholera, and anthrax. During the 20th and 21st centuries, effective vaccines were developed to prevent a wide range of diseases caused by viruses (e.g., chickenpox and shingles, hepatitis, measles, mumps, polio, and yellow fever) and bacteria (e.g., diphtheria, pneumococcal pneumonia, tetanus, and whooping cough).
Classes of Vaccines
For a vaccine to provide protection against a disease, it must expose an individual to pathogen-specific antigens that will stimulate a protective adaptive immune response. By its very nature, this entails some risk. As with any pharmaceutical drug, vaccines have the potential to cause adverse effects. However, the ideal vaccine causes no severe adverse effects and poses no risk of contracting the disease that it is intended to prevent. Various types of vaccines have been developed with these goals in mind. These different classes of vaccines are described in the next section and summarized in table 1.13.
Live Attenuated Vaccines
Live attenuated vaccines expose an individual to a weakened strain of a pathogen with the goal of establishing a subclinical infection that will activate the adaptive immune defenses. Pathogens are attenuated to decrease their virulence using methods such as genetic manipulation (to eliminate key virulence factors) or long-term culturing in an unnatural host or environment (to promote mutations and decrease virulence).
By establishing an active infection, live attenuated vaccines stimulate a more comprehensive immune response than some other types of vaccines. Live attenuated vaccines activate both cellular and humoral immunity and stimulate the development of memory for long-lasting immunity. In some cases, vaccination of one individual with a live attenuated pathogen can even lead to natural transmission of the attenuated pathogen to other individuals. This can cause the other individuals to also develop an active, subclinical infection that activates their adaptive immune defenses.
Disadvantages associated with live attenuated vaccines include the challenges associated with long-term storage and transport as well as the potential for a patient to develop signs and symptoms of disease during the active infection (particularly in immunocompromised patients). There is also a risk of the attenuated pathogen reverting back to full virulence. Table 1.13 lists examples of live attenuated vaccines.
Inactivated Vaccines
Inactivated vaccines contain whole pathogens that have been killed or inactivated with heat, chemicals, or radiation. For inactivated vaccines to be effective, the inactivation process must not affect the structure of key antigens on the pathogen.
Because the pathogen is killed or inactive, inactivated vaccines do not produce an active infection, and the resulting immune response is weaker and less comprehensive than that provoked by a live attenuated vaccine. Typically the response involves only humoral immunity, and the pathogen cannot be transmitted to other individuals. In addition, inactivated vaccines usually require higher doses and multiple boosters, possibly causing inflammatory reactions at the site of injection.
Despite these disadvantages, inactivated vaccines do have the advantages of long-term storage stability and ease of transport. Also, there is no risk of causing severe active infections. However, inactivated vaccines are not without their side effects. Table 1.13 lists examples of inactivated vaccines.
Subunit Vaccines
Whereas live attenuated and inactive vaccines expose an individual to a weakened or dead pathogen, subunit vaccines only expose the patient to the key antigens of a pathogen—not whole cells or viruses. Subunit vaccines can be produced either by chemically degrading a pathogen and isolating its key antigens or by producing the antigens through genetic engineering. Because these vaccines contain only the essential antigens of a pathogen, the risk of side effects is relatively low. Table 1.13 lists examples of subunit vaccines.
Toxoid Vaccines
Like subunit vaccines, toxoid vaccines do not introduce a whole pathogen to the patient; they contain inactivated bacterial toxins, called toxoids. Toxoid vaccines are used to prevent diseases in which bacterial toxins play an important role in pathogenesis. These vaccines activate humoral immunity that neutralizes the toxins. Table 1.13 lists examples of toxoid vaccines.
Conjugate Vaccines
A conjugate vaccine is a type of subunit vaccine that consists of a protein conjugated to a capsule polysaccharide. Conjugate vaccines have been developed to enhance the efficacy of subunit vaccines against pathogens that have protective polysaccharide capsules that help them evade phagocytosis, causing invasive infections that can lead to meningitis and other serious conditions. The subunit vaccines against these pathogens introduce T-independent capsular polysaccharide antigens that result in the production of antibodies that can opsonize the capsule and thus combat the infection; however, children under the age of two years do not respond effectively to these vaccines. Children do respond effectively when vaccinated with the conjugate vaccine, in which a protein with T-dependent antigens is conjugated to the capsule polysaccharide. The conjugated protein-polysaccharide antigen stimulates production of antibodies against both the protein and the capsule polysaccharide. Table 1.13 lists examples of conjugate vaccines.
| Class | Description | Advantages | Disadvantages | Examples |
|---|---|---|---|---|
| Live attenuated | Weakened strain of whole pathogen | Cellular and humoral immunity | Difficult to store and transport | Chickenpox, German measles, measles, mumps, tuberculosis, typhoid fever, yellow fever |
| Long-lasting immunity | Risk of infection in immunocompromised patients | |||
| Transmission to contacts | Risk of reversion | |||
| Inactivated | Whole pathogen killed or inactivated with heat, chemicals, or radiation | Ease of storage and transport | Weaker immunity (humoral only) | Cholera, hepatitis A, influenza, plague, rabies |
| No risk of severe active infection | Higher doses and more boosters required | |||
| Subunit | Immunogenic antigens | Lower risk of side effects | Limited longevity | Anthrax, hepatitis B, influenza, meningitis, papillomavirus, pneumococcal pneumonia, whooping cough |
| Multiple doses required | ||||
| No protection against antigenic variation | ||||
| Toxoid | Inactivated bacterial toxin | Humoral immunity to neutralize toxin | Does not prevent infection | Botulism, diphtheria, pertussis, tetanus |
| Conjugate | Capsule polysaccharide conjugated to protein | T-dependent response to capsule | Costly to produce | Meningitis (Haemophilus influenzae, Streptococcus pneumoniae, Neisseria meningitides) |
| Better response in young children | No protection against antigenic variation | |||
| May interfere with other vaccines |
Table 1.13: Classes of vaccines
1.13 Hypersensitivities
There are mechanisms by which adaptive immune defenses, both humoral and cellular, protect us from infectious diseases. However, these same protective immune defenses can also be responsible for undesirable reactions called hypersensitivity reactions. Hypersensitivity reactions are classified by their immune mechanism.
- Type I hypersensitivity reactions involve immunoglobulin E (IgE) antibody against soluble antigen, triggering mast cell degranulation.
- Type II hypersensitivity reactions involve IgG and IgM antibodies directed against cellular antigens, leading to cell damage mediated by other immune system effectors.
- Type III hypersensitivity reactions involve the interactions of IgG, IgM, and, occasionally, IgA[5] antibodies with antigen to form immune complexes. Accumulation of immune complexes in tissue leads to tissue damage mediated by other immune system effectors.
- Type IV hypersensitivity reactions are T-cell–mediated reactions that can involve tissue damage mediated by activated macrophages and cytotoxic T cells.
Type 1 Hypersensitivities
When a presensitized individual is exposed to an allergen, it can lead to a rapid immune response that occurs almost immediately. Such a response is called an allergy and is classified as a type I hypersensitivity. Allergens may be seemingly harmless substances such as animal dander, molds, or pollen. Allergens may also be substances considered innately more hazardous, such as insect venom or therapeutic drugs. Food intolerances can also yield allergic reactions as individuals become sensitized to foods such as peanuts or shellfish (figure 1.43). Regardless of the allergen, the first exposure activates a primary IgE antibody response that sensitizes an individual to type I hypersensitivity reaction upon subsequent exposure.
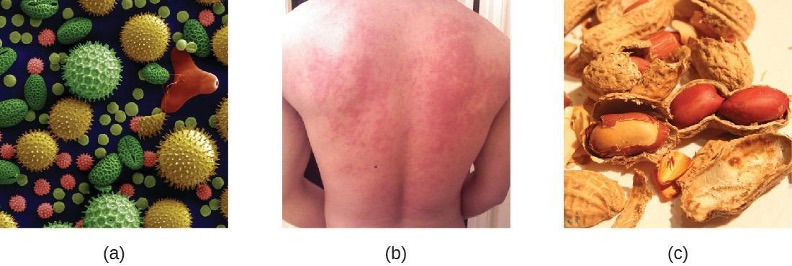
For susceptible individuals, a first exposure to an allergen activates a strong TH2 cell response (figure 1.44). Cytokines interleukin (IL)-4 and IL-13 from the TH2 cells activate B cells specific to the same allergen, resulting in clonal proliferation, differentiation into plasma cells, and antibody-class switching from production of IgM to production of IgE. The fragment crystallizable (Fc) regions of the IgE antibodies bind to specific receptors on the surface of mast cells throughout the body. It is estimated that each mast cell can bind up to 500,000 IgE molecules, with each IgE molecule having two allergen-specific fragment antigen-binding (Fab) sites available for binding allergen on subsequent exposures. By the time this occurs, the allergen is often no longer present and there is no allergic reaction, but the mast cells are primed for a subsequent exposure and the individual is sensitized to the allergen.
On subsequent exposure, allergens bind to multiple IgE molecules on mast cells, cross-linking the IgE molecules. Within minutes, this cross-linking of IgE activates the mast cells and triggers degranulation, a reaction in which the contents of the granules in the mast cell are released into the extracellular environment. Preformed components that are released from granules include histamine, serotonin, and bradykinin (table 1.14). The activated mast cells also release newly formed lipid mediators (leukotrienes and prostaglandins from membrane arachidonic acid metabolism) and cytokines such as tumor necrosis factor (table 1.15).
The chemical mediators released by mast cells collectively cause the inflammation and signs and symptoms associated with type I hypersensitivity reactions. Histamine stimulates mucus secretion in nasal passages and tear formation from lacrimal glands, promoting the runny nose and watery eyes of allergies. Interaction of histamine with nerve endings causes itching and sneezing. The vasodilation caused by several of the mediators can result in hives, headaches, angioedema (swelling that often affects the lips, throat, and tongue), and hypotension (low blood pressure). Bronchoconstriction caused by some of the chemical mediators leads to wheezing, dyspnea (difficulty breathing), coughing, and, in more severe cases, cyanosis (bluish color to the skin or mucous membranes). Vomiting can result from stimulation of the vomiting center in the cerebellum by histamine and serotonin. Histamine can also cause relaxation of intestinal smooth muscles and diarrhea.
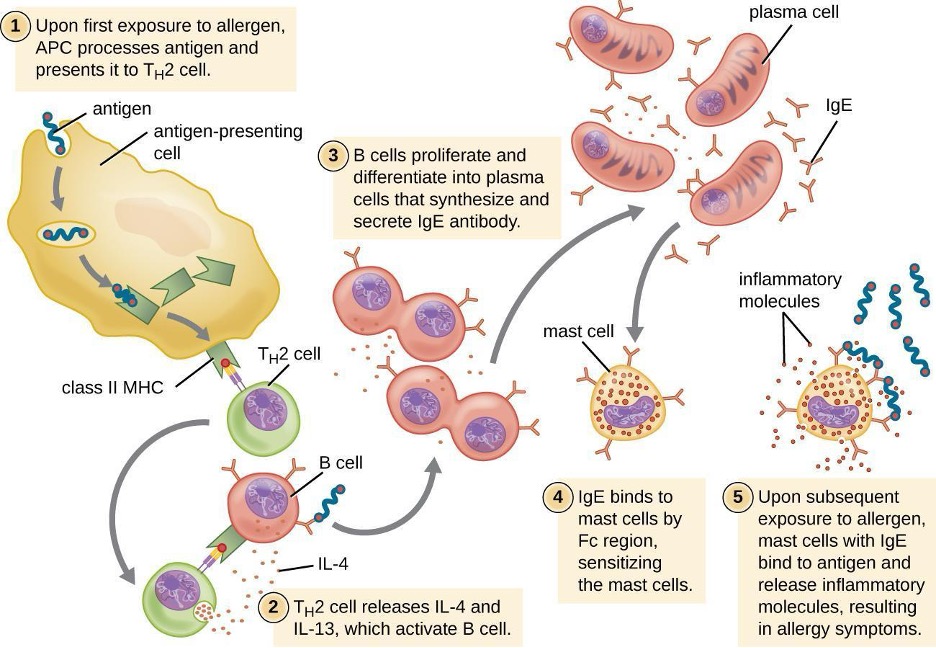
| Granule Component | Activity |
|---|---|
| Heparin | Stimulates the generation of bradykinin, which causes increased vascular permeability, vasodilation, bronchoconstriction, and increased mucus secretion |
| Histamine | Causes smooth-muscle contraction, increases vascular permeability, increases mucus and tear formation |
| Seratonin | Increases vascular permeability, causes vasodilation and smooth-muscle contraction |
Table 1.14: Selected preformed components of mast cell granules
| Chemical Mediator | Activity |
|---|---|
| Leukotriene | Causes smooth-muscle contraction and mucus secretion, increases vascular permeability |
| Prostaglandin | Causes smooth-muscle contraction and vasodilation |
| TNF-α (cytokine) | Causes inflammation and stimulates cytokine production by other cell types |
Table 1.15: Selected newly formed chemical mediators of inflammation and allergic response
Type I hypersensitivity reactions can be either localized or systemic. Localized type I hypersensitivity reactions include hay fever rhinitis, hives, and asthma (table 1.16). Systemic type I hypersensitivity reactions are referred to as anaphylaxis or anaphylactic shock. Although anaphylaxis shares many symptoms common with the localized type I hypersensitivity reactions, the swelling of the tongue and trachea, blockage of airways, dangerous drop in blood pressure, and development of shock can make anaphylaxis especially severe and life-threatening. In fact, death can occur within minutes of onset of signs and symptoms.
Late-phase reactions in type I hypersensitivities may develop 4–12 hours after the early phase and are mediated by eosinophils, neutrophils, and lymphocytes that have been recruited by chemotactic factors released from mast cells. Activation of these recruited cells leads to the release of more chemical mediators that cause tissue damage and late-phase symptoms of swelling and redness of the skin, coughing, wheezing, and nasal discharge.
Individuals who possess genes for maladaptive traits, such as intense type I hypersensitivity reactions to otherwise harmless components of the environment, would be expected to suffer reduced reproductive success. With this kind of evolutionary selective pressure, such traits would not be expected to persist in a population. This suggests that type I hypersensitivities may have an adaptive function. There is evidence that the IgE produced during type I hypersensitivity reactions is actually meant to counter helminth infections.[6] Helminths are one of few organisms that possess proteins that are targeted by IgE. In addition, there is evidence that helminth infections at a young age reduce the likelihood of type I hypersensitivities to innocuous substances later in life. Thus it may be that allergies are an unfortunate consequence of strong selection in the mammalian lineage or earlier for a defense against parasitic worms.
| Common Name | Cause | Signs and Symptoms |
|---|---|---|
| Allergy-induced asthma | Inhalation of allergens | Constriction of bronchi, labored breathing, coughing, chills, body aches |
| Anaphylaxis | Systemic reaction to allergens | Hives, itching, swelling of tongue and throat, nausea, vomiting, low blood pressure, shock |
| Hay fever | Inhalation of mold or pollen | Runny nose, watery eyes, sneezing |
| Hives (urticaria) | Food or drug allergens, insect stings | Raised, bumpy skin rash with itching; bumps may converge into large raised areas |
Table 1.16: Type I hypersensitivities
Type II (Cytotoxic) Hypersensitivities
Immune reactions categorized as type II hypersensitivities, or cytotoxic hypersensitivities, are mediated by IgG and IgM antibodies binding to cell-surface antigens or matrix-associated antigens on basement membranes. These antibodies can either activate complement, resulting in an inflammatory response and lysis of the targeted cells, or they can be involved in antibody-dependent cell-mediated cytotoxicity (ADCC) with cytotoxic T cells.
In some cases, the antigen may be a self-antigen, in which case the reaction would also be described as an autoimmune disease (for more information on autoimmune disorders, see section 1.14). In other cases, antibodies may bind to naturally occurring, but exogenous, cell-surface molecules such as antigens associated with blood typing found on red blood cells (RBCs). This leads to the coating of the RBCs by antibodies, activation of the complement cascade, and complement-mediated lysis of RBCs, as well as opsonization of RBCs for phagocytosis. Two examples of type II hypersensitivity reactions involving RBCs are hemolytic transfusion reaction (HTR) and hemolytic disease of the newborn (HDN). These type II hypersensitivity reactions, which will be discussed in greater detail, are summarized in table 1.17.
| Common Name | Cause | Signs and Symptoms |
|---|---|---|
| Hemolytic disease of the newborn (HDN) | IgG from mother crosses the placenta, targeting the fetus’ RBCs for destruction | Anemia, edema, enlarged liver or spleen, hydrops (fluid in body cavity), leading to death of newborn in severe cases |
| Hemolytic transfusion reactions (HTR) | IgG and IgM bind to antigens on transfused RBCs, targeting donor RBCs for destruction | Fever, jaundice, hypotension, disseminated intravascular coagulation, possibly leading to kidney failure and death |
Table 1.17: Common type II hypersensitivities
Immunohematology is the study of blood and blood-forming tissue in relation to the immune response. Antibody-initiated responses against blood cells are type II hypersensitivities, thus falling into the field of immunohematology. For students first learning about immunohematology, understanding the immunological mechanisms involved is made even more challenging by the complex nomenclature system used to identify different blood-group antigens, often called blood types. The first blood-group antigens either used alphabetical names or were named for the first person known to produce antibodies to the red blood cell antigen (e.g., Kell, Duffy, or Diego). However, in 1980, the International Society of Blood Transfusion (ISBT) Working Party on Terminology created a standard for blood-group terminology in an attempt to more consistently identify newly discovered blood group antigens. New antigens are now given a number and assigned to a blood-group system, collection, or series. However, even with this effort, blood-group nomenclature is still inconsistent.
ABO Blood Group Incompatibility
The recognition that individuals have different blood types was first described by Karl Landsteiner (1868–1943) in the early 1900s, based on his observation that serum from one person could cause a clumping of RBCs from another. These studies led Landsteiner to the identification of four distinct blood types. Subsequent research by other scientists determined that the four blood types were based on the presence or absence of surface carbohydrates A and B, and this provided the foundation for the ABO blood group system that is still in use today (figure 1.45). The functions of these antigens are unknown, but some have been associated with normal biochemical functions of the cell. Furthermore, ABO blood types are inherited as alleles (one from each parent), and they display patterns of dominant and codominant inheritance. The alleles for A and B blood types are codominant to each other, and both are dominant over blood type O. Therefore, individuals with genotypes of AA or AO have type A blood and express the A carbohydrate antigen on the surface of their RBCs. People with genotypes of BB or BO have type B blood and express the B carbohydrate antigen on the surface of their RBCs. Those with a genotype of AB have type AB blood and express both A and B carbohydrate antigens on the surface of their RBCs. Finally, individuals with a genotype of OO have type O blood and lack A and B carbohydrates on the surface of their RBCs.
It is important to note that the RBCs of all four ABO blood types share a common protein receptor molecule, and it is the addition of specific carbohydrates to the protein receptors that determines A, B, and AB blood types. The genes that are inherited for the A, B, and AB blood types encode enzymes that add the carbohydrate component to the protein receptor. Individuals with O blood type still have the protein receptor but lack the enzymes that would add carbohydrates that would make their red blood cell type A, B, or AB.
IgM antibodies in plasma that cross-react with blood group antigens not present on an individual’s own RBCs are called isohemagglutinins (figure 1.45). Isohemagglutinins are produced within the first few weeks after birth and persist throughout life. These antibodies are produced in response to exposure to environmental antigens from food and microorganisms. A person with type A blood has A antigens on the surface of their RBCs and will produce anti-B antibodies to environmental antigens that resemble the carbohydrate component of B antigens. Those with type B blood have B antigens on the surface of their RBCs and will produce anti-A antibodies to environmental antigens that are similar to the carbohydrate component of A antigens. People with blood type O lack both A and B antigens on their RBCs and, therefore, produce both anti-A and anti-B antibodies. Conversely, people with AB blood type have both A and B antigens on their RBCs and, therefore, lack anti-A and anti-B antibodies.
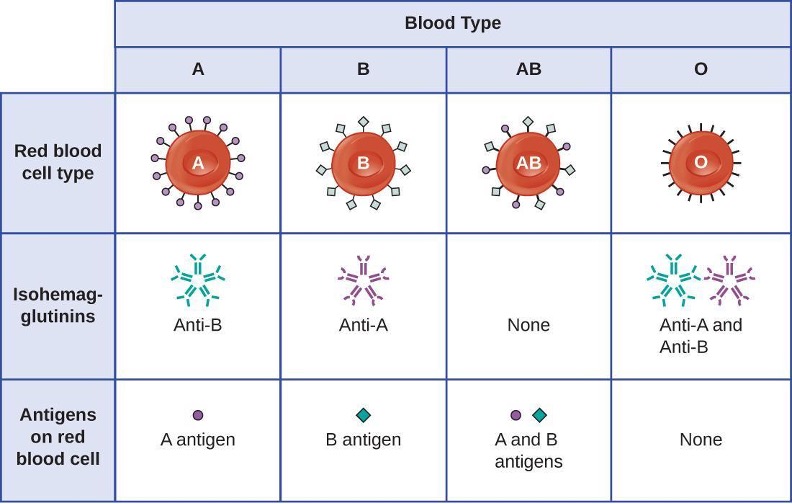
A patient may require a blood transfusion because they lack sufficient RBCs (anemia) or because they have experienced significant loss of blood volume through trauma or disease. Although the blood transfusion is given to help the patient, it is essential that the patient receive a transfusion with matching ABO blood type. A transfusion with an incompatible ABO blood type may lead to a strong, potentially lethal type II hypersensitivity cytotoxic response called hemolytic transfusion reaction (HTR) (figure 1.46).
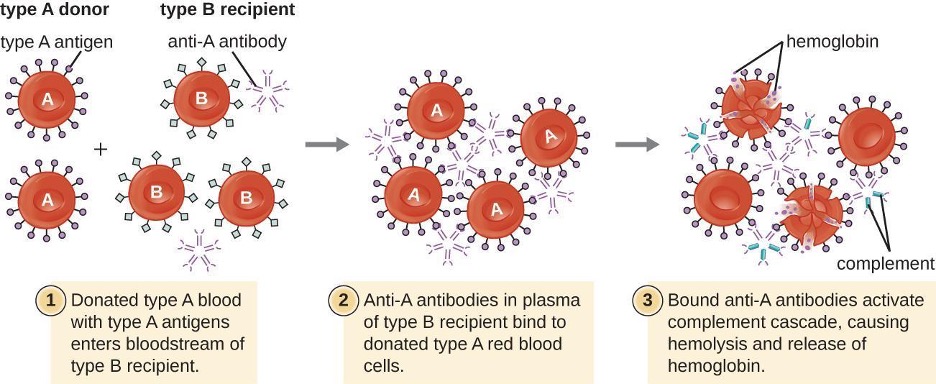
For instance, if a person with type B blood receives a transfusion of type A blood, their anti-A antibodies will bind to and agglutinate the transfused RBCs. In addition, activation of the classical complement cascade will lead to a strong inflammatory response, and the complement membrane attack complex (MAC) will mediate massive hemolysis of the transfused RBCs. The debris from damaged and destroyed RBCs can occlude blood vessels in the alveoli of the lungs and the glomeruli of the kidneys. Within 1 to 24 hours of an incompatible transfusion, the patient experiences fever, chills, pruritus (itching), urticaria (hives), dyspnea, hemoglobinuria (hemoglobin in the urine), and hypotension (low blood pressure). In the most serious reactions, dangerously low blood pressure can lead to shock, multi-organ failure, and death of the patient.
Hospitals, medical centers, and associated clinical laboratories typically use hemovigilance systems to minimize the risk of HTRs due to clerical error. Hemovigilance systems are procedures that track transfusion information from the donor source and blood products obtained to the follow-up of recipient patients. Hemovigilance systems used in many countries identify HTRs and their outcomes through mandatory reporting (e.g., to the Food and Drug Administration in the United States), and this information is valuable to help prevent such occurrences in the future. For example, if an HTR is found to be the result of laboratory or clerical error, additional blood products collected from the donor at that time can be located and labeled correctly to avoid additional HTRs. As a result of these measures, HTR-associated deaths in the United States occur in about one per 2 million transfused units.[7]
Rh Factors
Many different types of erythrocyte antigens have been discovered since the description of the ABO red cell antigens. The second most frequently described RBC antigens are Rh factors, named after the rhesus macaque (Macaca mulatta) factors identified by Karl Landsteiner and Alexander Weiner in 1940. The Rh system of RBC antigens is the most complex and immunogenic blood group system, with more than 50 specificities identified to date. Of all the Rh antigens, the one designated Rho (Weiner) or D (Fisher-Race) is the most immunogenic. Cells are classified as Rh positive (Rh+) if the Rho/D antigen is present or as Rh negative (Rh−) if the Rho/D antigen is absent. In contrast to the carbohydrate molecules that distinguish the ABO blood groups and are the targets of IgM isohemagglutinins in HTRs, the Rh factor antigens are proteins. As discussed in section 1.11, protein antigens activate B cells and antibody production through a T-cell–dependent mechanism, and the TH2 cells stimulate class switching from IgM to other antibody classes. In the case of Rh factor antigens, TH2 cells stimulate class switching to IgG, and this has important implications for the mechanism of HDN.
Like ABO incompatibilities, blood transfusions from a donor with the wrong Rh factor antigens can cause a type II hypersensitivity HTR. However, in contrast to the IgM isohemagglutinins produced early in life through exposure to environmental antigens, production of anti-Rh factor antibodies requires the exposure of an individual with Rh− blood to Rh+ positive RBCs and activation of a primary antibody response. Although this primary antibody response can cause an HTR in the transfusion patient, the hemolytic reaction would be delayed up to 2 weeks during the extended lag period of a primary antibody response (section 1.11). However, if the patient receives a subsequent transfusion with Rh+ RBCs, a more rapid HTR would occur with anti-Rh factor antibodies already present in the blood. Furthermore, the rapid secondary antibody response would provide even more anti-Rh factor antibodies for the HTR.
Rh factor incompatibility between mother and fetus can also cause a type II hypersensitivity hemolytic reaction, referred to as hemolytic disease of the newborn (HDN) (figure 1.47). If an Rh− woman carries an Rh+ baby to term, the mother’s immune system can be exposed to Rh+ fetal red blood cells. This exposure will usually occur during the last trimester of pregnancy and during the delivery process. If this exposure occurs, the Rh+ fetal RBCs will activate a primary adaptive immune response in the mother, and anti-Rh factor IgG antibodies will be produced. IgG antibodies are the only class of antibody that can cross the placenta from mother to fetus; however, in most cases, the first Rh+ baby is unaffected by these antibodies because the first exposure typically occurs late enough in the pregnancy that the mother does not have time to mount a sufficient primary antibody response before the baby is born.
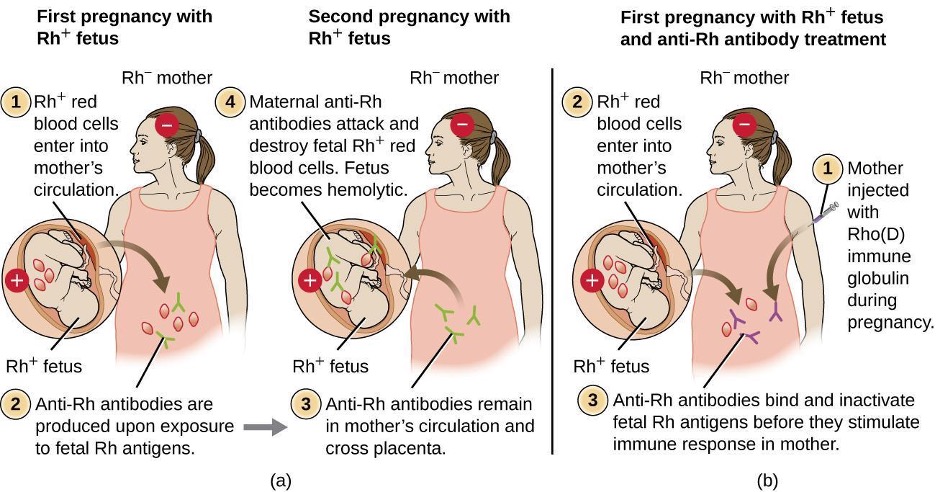
If a subsequent pregnancy with an Rh+ fetus occurs, however, the mother’s second exposure to the Rh factor antigens causes a strong secondary antibody response that produces larger quantities of anti-Rh factor IgG. These antibodies can cross the placenta from mother to fetus and cause HDN, a potentially lethal condition for the baby (figure 1.48).
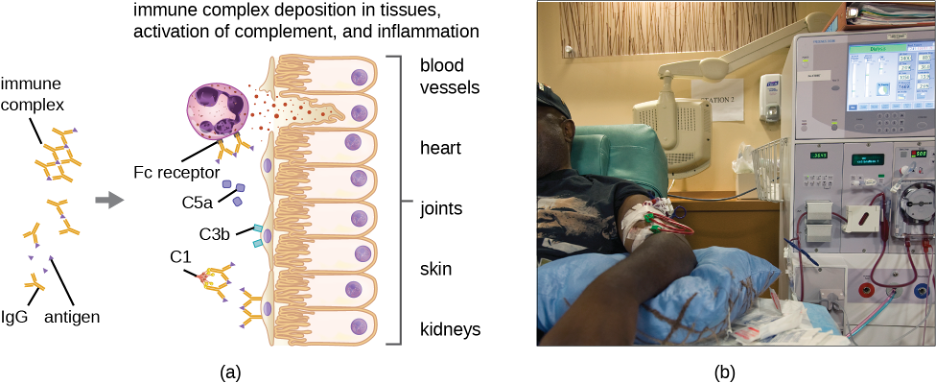
Prior to the development of techniques for diagnosis and prevention, Rh factor incompatibility was the most common cause of HDN, resulting in thousands of infant deaths each year worldwide.[8] For this reason, the Rh factors of prospective parents are regularly screened, and treatments have been developed to prevent HDN caused by Rh incompatibility. To prevent Rh factor-mediated HDN, human Rho(D) immune globulin (e.g., RhoGAM) is injected intravenously or intramuscularly into the mother during the 28th week of pregnancy and within 72 hours after delivery. Additional doses may be administered after events that may result in transplacental hemorrhage (e.g., umbilical blood sampling, chorionic villus sampling, abdominal trauma, amniocentesis). This treatment is initiated during the first pregnancy with an Rh+ fetus. The anti-Rh antibodies in Rho(D) immune globulin will bind to the Rh factor of any fetal RBCs that gain access to the mother’s bloodstream, preventing these Rh+ cells from activating the mother’s primary antibody response. Without a primary anti-Rh factor antibody response, the next pregnancy with an Rh+ will have minimal risk of HDN. However, the mother will need to be retreated with Rho(D) immune globulin during that pregnancy to prevent a primary anti-Rh antibody response that could threaten subsequent pregnancies.
Type III Hypersensitivities
Type III hypersensitivities are immune-complex reactions that were first characterized by Nicolas Maurice Arthus (1862–1945) in 1903. To produce antibodies for experimental procedures, Arthus immunized rabbits by injecting them with serum from horses. However, while immunizing rabbits repeatedly with horse serum, Arthus noticed a previously unreported and unexpected localized subcutaneous hemorrhage with edema at the site of injection. This reaction developed within 3 to 10 hours after injection. This localized reaction to non-self serum proteins was called an Arthus reaction. An Arthus reaction occurs when soluble antigens bind with IgG in a ratio that results in the accumulation of antigen-antibody aggregates called immune complexes.
A unique characteristic of type III hypersensitivity is antibody excess (primarily IgG), coupled with a relatively low concentration of antigen, resulting in the formation of small immune complexes that deposit on the surface of the epithelial cells lining the inner lumen of small blood vessels or on the surfaces of tissues (figure 1.48). This immune complex accumulation leads to a cascade of inflammatory events that include the following:
- IgG binding to antibody receptors on localized mast cells, resulting in mast-cell degranulation
- Complement activation with production of pro-inflammatory C3a and C5a (see section 1.4)
- Increased blood-vessel permeability with chemotactic recruitment of neutrophils and macrophages
Because these immune complexes are not an optimal size and are deposited on cell surfaces, they cannot be phagocytosed in the usual way by neutrophils and macrophages, which, in turn, are often described as “frustrated.” Although phagocytosis does not occur, neutrophil degranulation results in the release of lysosomal enzymes that cause extracellular destruction of the immune complex, damaging localized cells in the process. Activation of coagulation pathways also occurs, resulting in thrombi (blood clots) that occlude blood vessels and cause ischemia that can lead to vascular necrosis and localized hemorrhage.
Systemic type III hypersensitivity (serum sickness) occurs when immune complexes deposit in various body sites, resulting in a more generalized systemic inflammatory response. These immune complexes involve non-self proteins such as antibodies produced in animals for artificial passive immunity (see section 1.12 for more information on vaccines), certain drugs, or microbial antigens that are continuously released over time during chronic infections (e.g., subacute bacterial endocarditis, chronic viral hepatitis). The mechanisms of serum sickness are similar to those described in localized type III hypersensitivity but involve widespread activation of mast cells, complement, neutrophils, and macrophages, which causes tissue destruction in areas such as the kidneys, joints, and blood vessels. As a result of tissue destruction, symptoms of serum sickness include chills, fever, rash, vasculitis, and arthritis. Development of glomerulonephritis or hepatitis is also possible.
Autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis can also involve damaging type III hypersensitivity reactions when auto-antibodies form immune complexes with self antigens. These conditions are discussed in section 1.14.
Type IV Hypersensitivities
Type IV hypersensitivities are not mediated by antibodies like the other three types of hypersensitivities. Rather, type IV hypersensitivities are regulated by T cells and involve the action of effector cells. These types of hypersensitivities can be organized into three subcategories based on T-cell subtype, type of antigen, and the resulting effector mechanism (table 1.18).
In the first type IV subcategory, CD4 TH1-mediated reactions are described as delayed-type hypersensitivities (DTH). The sensitization step involves the introduction of antigen into the skin and phagocytosis by local antigen presenting cells (APCs). The APCs activate helper T cells, stimulating clonal proliferation and differentiation into memory TH1 cells. Upon subsequent exposure to the antigen, these sensitized memory TH1 cells release cytokines that activate macrophages, and activated macrophages are responsible for much of the tissue damage. Examples of this TH1-mediated hypersensitivity are observed in the Mantoux tuberculin skin test and contact dermatitis.
In the second type IV subcategory, CD4 TH2-mediated reactions result in chronic asthma or chronic allergic rhinitis. In these cases, the soluble antigen is first inhaled, resulting in eosinophil recruitment and activation with the release of cytokines and inflammatory mediators.
In the third type IV subcategory, CD8 cytotoxic T lymphocyte (CTL)-mediated reactions are associated with tissue transplant rejection and contact dermatitis (figure 1.49). For this form of cell-mediated hypersensitivity, APCs process and present the antigen with MHC I to naïve CD8 T cells. When these naïve CD8 T cells are activated, they proliferate and differentiate into CTLs. Activated TH1 cells can also enhance the activation of the CTLs. The activated CTLs then target and induce granzyme-mediated apoptosis in cells presenting the same antigen with MHC I. These target cells could be “self” cells that have absorbed the foreign antigen (such as with contact dermatitis due to poison ivy), or they could be transplanted tissue cells displaying foreign antigen from the donor.
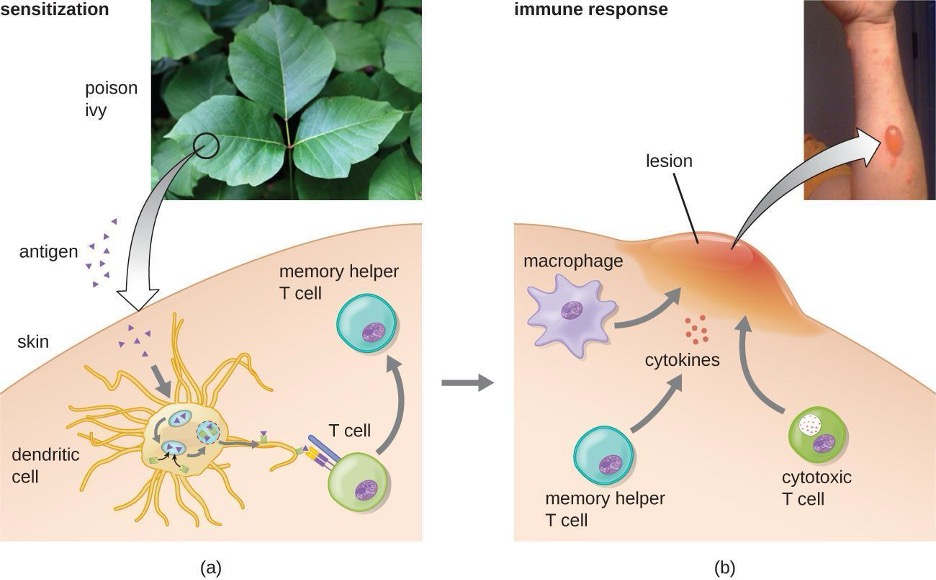
| Subcategory | Antigen | Effector Mechanism | Examples |
|---|---|---|---|
| 1 | Soluble antigen | Activated macrophages damage tissue and promote inflammatory response | Contact dermatitis (e.g., exposure to latex) and delayed-type hypersensitivity (e.g., tuberculin reaction) |
| 2 | Soluble antigen | Eosinophil recruitment and activation release cytokines and pro-inflammatory chemicals | Chronic asthma and chronic allergic rhinitis |
| 3 | Cell-associated antigen | CTL-mediated cytotoxicity | Contact dermatitis (e.g., contact with poison ivy) and tissue-transplant rejection |
Table 1.18: Type IV hypersensitivities
Hypersensitivity Pneumonitis
Some diseases caused by hypersensitivities are not caused exclusively by one type. For example, hypersensitivity pneumonitis (HP), which is often an occupational or environmental disease, occurs when the lungs become inflamed due to an allergic reaction to inhaled dust, endospores, bird feathers, bird droppings, molds, or chemicals. HP goes by many different names associated with various forms of exposure (figure 1.50). HP associated with bird droppings is sometimes called pigeon fancier’s lung or poultry worker’s lung—both common in bird breeders and handlers. Cheese handler’s disease, farmer’s lung, sauna takers’ disease, and hot-tub lung are other names for HP associated with exposure to molds in various environments.

Pathology associated with HP can be due to both type III (mediated by immune complexes) and type IV (mediated by TH1 cells and macrophages) hypersensitivities. Repeated exposure to allergens can cause alveolitis due to the formation of immune complexes in the alveolar wall of the lung accompanied by fluid accumulation, and the formation of granulomas and other lesions in the lung as a result of TH1-mediated macrophage activation. Alveolitis with fluid and granuloma formation results in poor oxygen perfusion in the alveoli, which, in turn, can cause symptoms such as coughing, dyspnea, chills, fever, sweating, myalgias, headache, and nausea. Symptoms may occur as quickly as 2 hours after exposure and can persist for weeks if left untreated.
Table 1.19 summarizes the mechanisms and effects of each type of hypersensitivity discussed in this section.
| Type I | Type II | Type III | Type IV | |
|---|---|---|---|---|
| Immune Reactant | IgE | IgG or IgM | IgG and IgM | T cells |
| Antigen form | Soluble antigen | Cell-bound antigen | Soluble antigen | Soluble or cell-bound antigen |
| Mechanism of activation | Allergen-specific IgE antibodies bind to mast cells via their Fc receptor. When the specific allergen binds to the IgE, cross-linking of IgE induces degranulation of mast cells. | IgG or IgM antibody binds to cellular antigen, leading to complement activation and cell lysis. IgG can also mediate ADCC with cytotoxic T cells, natural killer cells, macrophages, and neutrophils. | Antigen-antibody complexes are deposited in tissues. Complement activation provides inflammatory mediators and recruits neutrophils. Enzymes released from neutrophils damage tissue. | TH1 cells secrete cytokines, which activate macrophages and cytotoxic T cells. |
| Examples of hypersensitivity reactions | Local and systemic anaphylaxis, seasonal hay fever, food allergies, and drug allergies | Red blood cell destruction after transfusion with mismatched blood types or during hemolytic disease of the newborn. | Local and systemic Post-streptococcal glomerulonephritis, rheumatoid arthritis, and systemic lupus erythematosus | Contact dermatitis, type I diabetes mellitus, and multiple sclerosis |
Table 1.19: Components of the immune system cause four types of hypersensitivities. Notice that types I–III are B-cell/antibody-mediated hypersensitivities, whereas type IV hypersensitivity is exclusively a T-cell phenomenon.
Diagnosis of Hypersensitivities
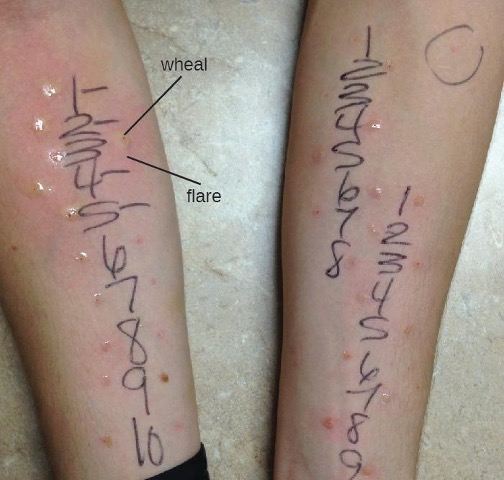
Diagnosis of type I hypersensitivities is a complex process requiring several diagnostic tests in addition to a well-documented patient history. Serum IgE levels can be measured, but elevated IgE alone does not confirm allergic disease. As part of the process to identify the antigens responsible for a type I reaction allergy, testing through a prick puncture skin test (PPST) or an intradermal test can be performed. PPST is carried out with the introduction of allergens in a series of superficial skin pricks on the patient’s back or arms (figure 1.51). PPSTs are considered to be the most convenient and least expensive way to diagnose allergies, according to the US Joint Council of Allergy and the European Academy of Allergy and Immunology. The second type of testing, the intradermal test, requires injection into the dermis with a small needle. This needle, also known as a tuberculin needle, is attached to a syringe containing a small amount of allergen. Both the PPST and the intradermal tests are observed for 15–20 minutes for a wheal-flare reaction to the allergens. Measurement of any wheal (a raised, itchy bump) and flare (redness) within minutes indicates a type I hypersensitivity, and the larger the wheal-flare reaction, the greater the patient’s sensitivity to the allergen.
Type III hypersensitivities can often be misdiagnosed because of their nonspecific inflammatory nature. The symptoms are easily visible, but they may be associated with any of a number of other diseases. A strong, comprehensive patient history is crucial to proper and accurate diagnosis. Tests used to establish the diagnosis of hypersensitivity pneumonitis (resulting from type III hypersensitivity) include bronchoalveolar lavage (BAL), pulmonary function tests, and high-resolution computed tomography (HRCT).
Treatments of Hypersensitivities
Allergic reactions can be treated in various ways. Prevention of allergic reactions can be achieved by desensitization (hyposensitization) therapy, which can be used to reduce the hypersensitivity reaction through repeated injections of allergens. Extremely dilute concentrations of known allergens (determined from the allergen tests) are injected into the patient at prescribed intervals (e.g., weekly). The quantity of allergen delivered by the shots is slowly increased over a buildup period until an effective dose is determined and that dose is maintained for the duration of treatment, which can last years. Patients are usually encouraged to remain in the doctor’s office for 30 minutes after receiving the injection in case the allergens administered cause a severe systemic reaction. Doctors’ offices that administer desensitization therapy must be prepared to provide resuscitation and drug treatment in the case of such an event.
Desensitization therapy is used for insect sting allergies and environmental allergies. The allergy shots elicit the production of different interleukins and IgG antibody responses instead of IgE. When excess allergen-specific IgG antibodies are produced and bind to the allergen, they can act as blocking antibodies to neutralize the allergen before it can bind IgE on mast cells. There are early studies using oral therapy for desensitization of food allergies that are promising.[9][10] These studies involve feeding children who have allergies tiny amounts of the allergen (e.g., peanut flour) or related proteins over time. Many of the subjects show reduced severity of reaction to the food allergen after the therapy.
There are also therapies designed to treat severe allergic reactions. Emergency systemic anaphylaxis is treated initially with an epinephrine injection, which can counteract the drop in blood pressure. Individuals with known severe allergies often carry a self-administering auto-injector that can be used in case of exposure to the allergen (e.g., an insect sting or accidental ingestion of a food that causes a severe reaction). By self-administering an epinephrine shot (or sometimes two), the patient can stem the reaction long enough to seek medical attention. Follow-up treatment generally involves giving the patient antihistamines and slow-acting corticosteroids for several days after the reaction to prevent potential late-phase reactions. However, the effects of antihistamine and corticosteroid treatment are not well studied and are used based on theoretical considerations.
Treatment of milder allergic reactions typically involves antihistamines and other anti-inflammatory drugs. A variety of antihistamine drugs are available, in both prescription and over-the-counter strengths. There are also antileukotriene and antiprostaglandin drugs that can be used in tandem with antihistamine drugs in a combined (and more effective) therapy regime.
Treatments of type III hypersensitivities include preventing further exposure to the antigen and the use of anti-inflammatory drugs. Some conditions can be resolved when exposure to the antigen is prevented. Anti-inflammatory corticosteroid inhalers can also be used to diminish inflammation to allow lung lesions to heal. Systemic corticosteroid treatment, oral or intravenous, is also common for type III hypersensitivities affecting body systems. Treatment of hypersensitivity pneumonitis includes avoiding the allergen, along with the possible addition of prescription steroids such as prednisone to reduce inflammation.
Treatment of type IV hypersensitivities includes antihistamines, anti-inflammatory drugs, analgesics, and, if possible, eliminating further exposure to the antigen.
1.14 Autoimmune Disorders
In 1970, artist Walt Kelly developed a poster promoting Earth Day, featuring a character from Pogo, his daily newspaper comic strip. In the poster, Pogo looks out across a litter-strewn forest and says wryly, “We have met the enemy and he is us.” Pogo was not talking about the human immune system, but he very well could have been. Although the immune system protects the body by attacking invading “enemies” (pathogens), in some cases, the immune system can mistakenly identify the body’s own cells as the enemy, resulting in autoimmune disease.
Autoimmune diseases are those in which the body is attacked by its own specific adaptive immune response. In normal, healthy states, the immune system induces tolerance, which is a lack of an anti-self immune response. However, with autoimmunity, there is a loss of immune tolerance, and the mechanisms responsible for autoimmune diseases include type II, III, and IV hypersensitivity reactions. Autoimmune diseases can have a variety of mixed symptoms that flare up and disappear, making diagnosis difficult.
The causes of autoimmune disease are a combination of the individual’s genetic makeup and the effect of environmental influences, such as sunlight, infections, medications, and environmental chemicals. However, the vagueness of this list reflects our poor understanding of the etiology of these diseases. Except in a very few specific diseases, the initiation event(s) of most autoimmune states has not been fully characterized.
There are several possible causes for the origin of autoimmune diseases and autoimmunity is likely due to several factors. Evidence now suggests that regulatory T and B cells play an essential role in the maintenance of tolerance and prevention of autoimmune responses. The regulatory T cells are especially important for inhibiting autoreactive T cells that are not eliminated during thymic selection and escape the thymus (see section 1.10). In addition, antigen mimicry between pathogen antigens and our own self antigens can lead to cross-reactivity and autoimmunity. Hidden self-antigens may become exposed because of trauma, drug interactions, or disease states, and trigger an autoimmune response. All of these factors could contribute to autoimmunity. Ultimately, damage to tissues and organs in the autoimmune disease state comes as a result of inflammatory responses that are inappropriate; therefore, treatment often includes immunosuppressive drugs and corticosteroids.
Organ-Specific Autoimmune Diseases
Some autoimmune diseases are considered organ specific, meaning that the immune system targets specific organs or tissues. Examples of organ-specific autoimmune diseases include celiac disease, Graves disease, Hashimoto thyroiditis, type I diabetes mellitus, and Addison disease.
Celiac Disease
Celiac disease is largely a disease of the small intestine, although other organs may be affected. People in their 30s and 40s, and children are most commonly affected, but celiac disease can begin at any age. It results from a reaction to proteins, commonly called gluten, found mainly in wheat, barley, rye, and some other grains. The disease has several genetic causes (predispositions) and poorly understood environmental influences. On exposure to gluten, the body produces various autoantibodies and an inflammatory response. The inflammatory response in the small intestine leads to a reduction in the depth of the microvilli of the mucosa, which hinders absorption and can lead to weight loss and anemia. The disease is also characterized by diarrhea and abdominal pain, symptoms that are often misdiagnosed as irritable bowel syndrome.
Diagnosis of celiac disease is accomplished from serological tests for the presence of primarily IgA antibodies to components of gluten, the transglutaminase enzyme, and autoantibodies to endomysium, a connective tissue surrounding muscle fibers. Serological tests are typically followed up with endoscopy and biopsy of the duodenal mucosa. Serological screening surveys have found about 1% of individuals in the United Kingdom are positive even though they do not all display symptoms.[11] This early recognition allows for more careful monitoring and prevention of severe disease.
Celiac disease is treated with complete removal of gluten-containing foods from the diet, which results in improved symptoms and reduced risk of complications. Other theoretical approaches include breeding grains that do not contain the immunologically reactive components or developing dietary supplements that contain enzymes that break down the protein components that cause the immune response.[12]
Disorders of the Thyroid
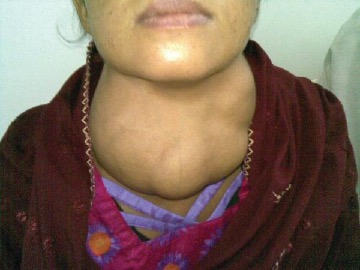
Graves disease is the most common cause of hyperthyroidism in the United States. Symptoms of Graves disease result from the production of thyroid-stimulating immunoglobulin (TSI), also called TSH-receptor antibody. TSI targets and binds to the receptor for thyroid stimulating hormone (TSH), which is naturally produced by the pituitary gland. TSI may cause conflicting symptoms because it may stimulate the thyroid to make too much thyroid hormone or block thyroid hormone production entirely, making diagnosis more difficult. Signs and symptoms of Graves disease include heat intolerance, rapid and irregular heartbeat, weight loss, goiter (a swollen thyroid gland, protruding under the skin of the throat [figure 1.52]) and exophthalmia (bulging eyes) often referred to as Graves ophthalmopathy (figure 1.53).
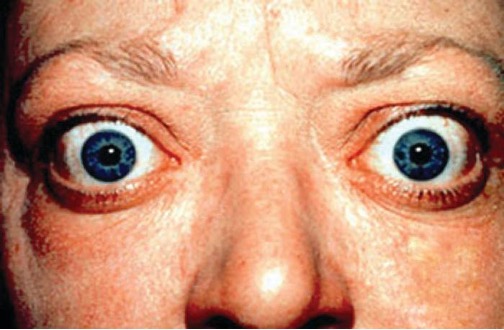
The most common cause of hypothyroidism in the United States is Hashimoto thyroiditis, also called chronic lymphocytic thyroiditis. Patients with Hashimoto thyroiditis often develop a spectrum of different diseases because they are more likely to develop additional autoimmune diseases such as Addison disease (discussed later in this section), type 1 diabetes, rheumatoid arthritis, and celiac disease. Hashimoto thyroiditis is a TH1 cell-mediated disease that occurs when the thyroid gland is attacked by cytotoxic lymphocytes, macrophages, and autoantibodies. This autoimmune response leads to numerous symptoms that include goiter (figure 1.52), cold intolerance, muscle weakness, painful and stiff joints, depression, and memory loss.
Type 1 Diabetes
Juvenile diabetes, or type 1 diabetes mellitus, is usually diagnosed in children and young adults. It is a T-cell-dependent autoimmune disease characterized by the selective destruction of the β cells of the islets of Langerhans in the pancreas by CD4 TH1-mediated CD8 T cells, anti-β-cell antibodies, and macrophage activity. There is also evidence that viral infections can have either a potentiating or inhibitory role in the development of type 1 diabetes (T1D) mellitus. The destruction of the β cells causes a lack of insulin production by the pancreas. In T1D, β-cell destruction may take place over several years, but symptoms of hyperglycemia, extreme increase in thirst and urination, weight loss, and extreme fatigue usually have a sudden onset, and diagnosis usually does not occur until most β cells have already been destroyed.
Autoimmune Addison Disease
Destruction of the adrenal glands (the glands lying above the kidneys that produce glucocorticoids, mineralocorticoids, and sex steroids) is the cause of Addison disease, also called primary adrenal insufficiency (PAI). Today, up to 80% of Addison disease cases are diagnosed as autoimmune Addison disease (AAD), which is caused by an autoimmune response to adrenal tissues disrupting adrenal function. Disruption of adrenal function causes impaired metabolic processes that require normal steroid hormone levels, causing signs and symptoms throughout the body. There is evidence that both humoral and CD4 TH1-driven CD8 T-cell–mediated immune mechanisms are directed at the adrenal cortex in AAD. There is also evidence that the autoimmune response is associated with autoimmune destruction of other endocrine glands as well, such as the pancreas and thyroid, conditions collectively referred to as autoimmune polyendocrine syndromes (APS). In up to 80% of patients with AAD, antibodies are produced to three enzymes involved in steroid synthesis: 21-hydroxylase (21-OH), 17α-hydroxylase, and cholesterol side-chain–cleaving enzyme.[13] The most common autoantibody found in AAD is to 21-OH, and antibodies to any of the key enzymes for steroid production are diagnostic for AAD. The adrenal cortex cells are targeted, destroyed, and replaced with fibrous tissue by immune-mediated inflammation. In some patients, at least 90% of the adrenal cortex is destroyed before symptoms become diagnostic.
Symptoms of AAD include weakness, nausea, decreased appetite, weight loss, hyperpigmentation (figure 1.54), hyperkalemia (elevated blood potassium levels), hyponatremia (decreased blood sodium levels), hypoglycemia (decreased levels of blood sugar), hypotension (decreased blood pressure), anemia, lymphocytosis (decreased levels of white blood cells), and fatigue. Under extreme stress, such as surgery, accidental trauma, or infection, patients with AAD may experience an adrenal crisis that causes the patient to vomit, experience abdominal pain, back or leg cramps, and even severe hypotension leading to shock.
Systemic Autoimmune Diseases
Whereas organ-specific autoimmune diseases target specific organs or tissues, systemic autoimmune diseases are more generalized, targeting multiple organs or tissues throughout the body. Examples of systemic autoimmune diseases include multiple sclerosis, myasthenia gravis, psoriasis, rheumatoid arthritis, and systemic lupus erythematosus.
Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune central nervous system disease that affects the brain and spinal cord. Lesions in multiple locations within the central nervous system are a hallmark of multiple sclerosis and are caused by infiltration of immune cells across the blood-brain barrier. The immune cells include T cells that promote inflammation, demyelination, and neuron degeneration, all of which disrupt neuronal signaling. Symptoms of MS include visual disturbances; muscle weakness; difficulty with coordination and balance; sensations such as numbness, prickling, or “pins and needles”; and cognitive and memory problems.
Myasthenia Gravis
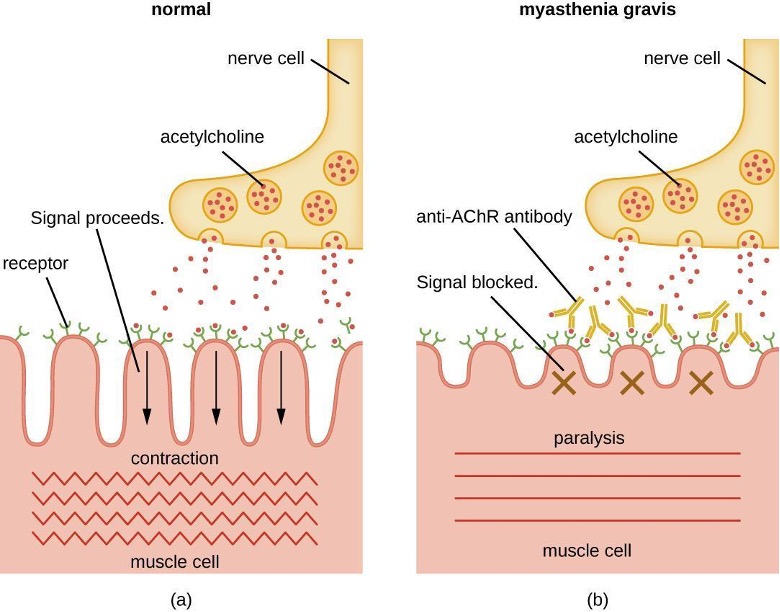
Autoantibodies directed against acetylcholine receptors (AChRs) in the synaptic cleft of neuromuscular junctions lead to myasthenia gravis (figure 1.55). Anti-AChR antibodies are high-affinity IgGs and their synthesis requires activated CD4 T cells to interact with and stimulate B cells. Once produced, the anti-AChR antibodies affect neuromuscular transmission by at least three mechanisms:
- Complement binding and activation at the neuromuscular junction
- Accelerated AChR endocytosis of molecules cross-linked by antibodies
- Functional AChR blocking, which prevents normal acetylcholine attachment to, and activation of, AChR
Regardless of the mechanism, the effect of anti-AChR is extreme muscle weakness and potentially death through respiratory arrest in severe cases.
Psoriasis
Psoriasis is a skin disease that causes itchy or sore patches of thick, red skin with silvery scales on the elbows, knees, scalp, back, face, palms, feet, and sometimes other areas. Some individuals with psoriasis also get a form of arthritis called psoriatic arthritis, in which the joints can become inflamed. Psoriasis results from the complex interplay between keratinocytes, dendritic cells, and T cells, and the cytokines produced by these various cells. In a process called cell turnover, skin cells that grow deep in the skin rise to the surface. Normally, this process takes a month. In psoriasis, as a result of cytokine activation, cell turnover happens in just a few days. The thick inflamed patches of skin that are characteristic of psoriasis develop because the skin cells rise too fast.
Rheumatoid Arthritis
The most common chronic inflammatory joint disease is rheumatoid arthritis (RA) (figure 1.56) and it is still a major medical challenge because of unsolved questions related to the environmental and genetic causes of the disease. RA involves type III hypersensitivity reactions and the activation of CD4 T cells, resulting in chronic release of the inflammatory cytokines IL-1, IL-6, and tumor necrosis factor-α (TNF-α). The activated CD4 T cells also stimulate the production of rheumatoid factor (RF) antibodies and anticyclic citrullinated peptide antibodies (anti-CCP) that form immune complexes. Increased levels of acute-phase proteins, such as C-reactive protein (CRP), are also produced as part of the inflammatory process and participate in complement fixation with the antibodies on the immune complexes. The formation of immune complexes and reaction to the immune factors cause an inflammatory process in joints, particularly in the hands, feet, and legs. Diagnosis of RA is based on elevated levels of RF, anti-CCP, quantitative CRP, and the erythrocyte sedimentation rate (ESR) (modified Westergren). In addition, radiographs, ultrasound, or magnetic resonance imaging scans can identify joint damage, such as erosions, a loss of bone within the joint, and narrowing of joint space.
Systemic Lupus Erythematosus
The damage and pathology of systemic lupus erythematosus (SLE) is caused by type III hypersensitivity reactions. Autoantibodies produced in SLE are directed against nuclear and cytoplasmic proteins. Anti-nuclear antibodies (ANAs) are present in more than 95% of patients with SLE,[14] with additional autoantibodies including anti-double–stranded DNA (ds-DNA) and anti-Sm antibodies (antibodies to small nuclear ribonucleoprotein). Anti-ds-DNA and anti-Sm antibodies are unique to patients with SLE; thus, their presence is included in the classification criteria of SLE. Cellular interaction with autoantibodies leads to nuclear and cellular destruction, with components released after cell death leading to the formation of immune complexes.
Because autoantibodies in SLE can target a wide variety of cells, symptoms of SLE can occur in many body locations. However, the most common symptoms include fatigue, fever with no other cause, hair loss, and a sunlight-sensitive “butterfly” or wolf-mask (lupus) rash that is found in about 50% of people with SLE (figure 1.57). The rash is most often seen over the cheeks and bridge of the nose, but can be widespread. Other symptoms may appear depending on affected areas. The joints may be affected, leading to arthritis of the fingers, hands, wrists, and knees. Effects on the brain and nervous system can lead to headaches, numbness, tingling, seizures, vision problems, and personality changes. There may also be abdominal pain, nausea, vomiting, arrhythmias, shortness of breath, and blood in the sputum. Effects on the skin can lead to additional areas of skin lesions, and vasoconstriction can cause color changes in the fingers when they are cold (Raynaud phenomenon). Effects on the kidneys can lead to edema in the legs and weight gain. A diagnosis of SLE depends on identification of four of 11 of the most common symptoms and confirmed production of an array of autoantibodies unique to SLE. A positive test for ANAs alone is not diagnostic.
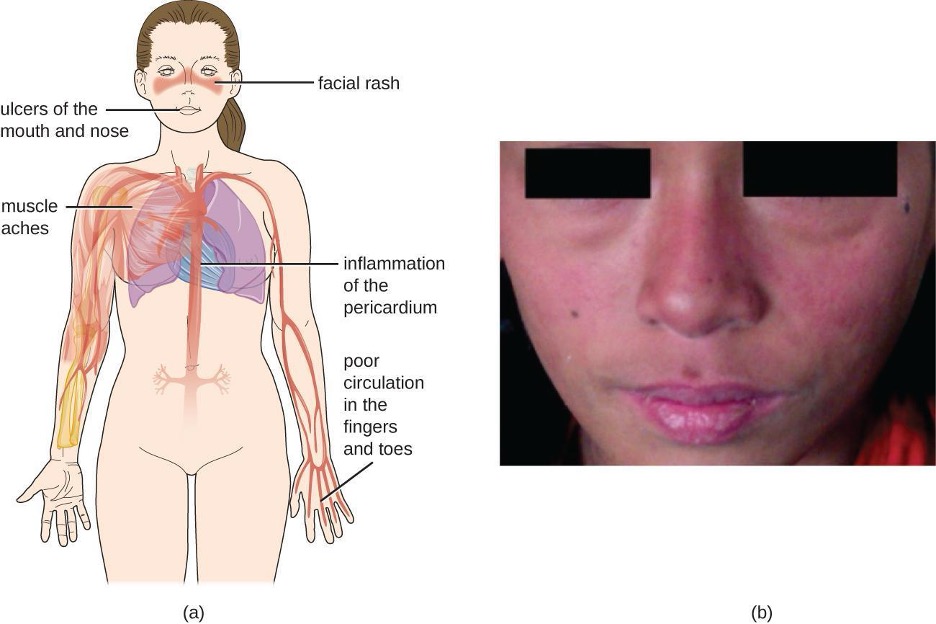
Table 1.20 summarizes the causes, signs, and symptoms of select autoimmune diseases.
| Disease | Cause | Signs and Symptoms |
|---|---|---|
| Addison disease | Destruction of adrenal gland cells by cytotoxic T cells | Weakness, nausea, hypotension, fatigue; adrenal crisis with severe pain in abdomen, lower back, and legs; circulatory system collapse, kidney failure |
| Celiac disease | Antibodies to gluten become autoantibodies that target cells of the small intestine | Severe diarrhea, abdominal pain, anemia, malnutrition |
| Diabetes mellitus (type I) | Cytotoxic T-cell destruction of the insulin-producing β cells of the pancreas | Hyperglycemia, extreme increase in thirst and urination, weight loss, extreme fatigue |
| Graves disease | Autoantibodies target thyroid-stimulating hormone receptors, resulting in overstimulation of the thyroid | Hyperthyroidism with rapid and irregular heartbeat, heat intolerance, weight loss, goiter, exophthalmia |
| Hashimoto thyroiditis | Thyroid gland is attacked by cytotoxic T cells, lymphocytes, macrophages, and autoantibodies | Thyroiditis with goiter, cold intolerance, muscle weakness, painful and stiff joints, depression, memory loss |
| Multiple sclerosis (MS) | Cytotoxic T-cell destruction of the myelin sheath surrounding nerve axons in the central nervous system | Visual disturbances, muscle weakness, impaired coordination and balance, numbness, prickling or “pins and needles” sensations, impaired cognitive function and memory |
| Myasthenia gravis | Autoantibodies directed against acetylcholine receptors within the neuromuscular junction | Extreme muscle weakness eventually leading to fatal respiratory arrest |
| Psoriasis | Cytokine activation of keratinocytes causes rapid and excessive epidermal cell turnover | Itchy or sore patches of thick, red skin with silvery scales; commonly affects elbows, knees, scalp, back, face, palms, feet |
| Rheumatoid arthritis | Autoantibodies, immune complexes, complement activation, phagocytes, and T cells damage membranes and bone in joints | Joint inflammation, pain and disfigurement, chronic systemic inflammation |
| Systemic lupus erythematosus (SLE) | Autoantibodies directed against nuclear and cytoplasmic molecules form immune complexes that deposit in tissues. Phagocytic cells and complement activation cause tissue damage and inflammation | Fatigue, fever, joint pain and swelling, hair loss, anemia, clotting, a sunlight-sensitive "butterfly" rash, skin lesions, photosensitivity, decreased kidney function, memory loss, confusion, depression |
Table 1.20: Select autoimmune diseases
1.15 Organ Transplantation and Rejection
A graft is the transplantation of an organ or tissue to a different location, with the goal of replacing a missing or damaged organ or tissue. Grafts are typically relocated without their attachments to the circulatory system and must reestablish these, in addition to the other connections and interactions with their new surrounding tissues. There are different types of grafts depending on the source of the new tissue or organ. Tissues that are transplanted from one genetically distinct individual to another within the same species are called allografts. An interesting variant of the allograft is an isograft, in which tissue from one twin is transplanted to another. As long as the twins are monozygotic (therefore, essentially genetically identical), the transplanted tissue is virtually never rejected. If tissues are transplanted from one area on an individual to another area on the same individual (e.g., a skin graft on a burn patient), it is known as an autograft. If tissues from an animal are transplanted into a human, this is called a xenograft.
Transplant Rejection
The different types of grafts described above have varying risks for rejection (table 1.21). Rejection occurs when the recipient’s immune system recognizes the donor tissue as foreign (non-self), triggering an immune response. The major histocompatibility complex markers MHC I and MHC II, more specifically identified as human leukocyte antigens (HLAs), play a role in transplant rejection. The HLAs expressed in tissue transplanted from a genetically different individual or species may be recognized as non-self molecules by the host’s dendritic cells. If this occurs, the dendritic cells will process and present the foreign HLAs to the host’s helper T cells and cytotoxic T cells, thereby activating them. Cytotoxic T cells then target and kill the grafted cells through the same mechanism they use to kill virus-infected cells; helper T cells may also release cytokines that activate macrophages to kill graft cells.
| Graft | Procedure | Complications |
|---|---|---|
| Autograft | From self to self | No rejection concerns |
| Isograft | from identical twin to twin | Little concern of rejection |
| Allograft | from relative or nonrelative to individual | Rejection possible |
| Xenograft | From animal to human | Rejection possible |
Table 1.21: Types of tissue and organ grafts and their complications
With the three highly polymorphic MHC I genes in humans (HLA-A, HLA-B, and HLA-C) determining compatibility, each with many alleles segregating in a population, odds are extremely low that a randomly chosen donor will match a recipient’s six-allele genotype (the two alleles at each locus are expressed codominantly). This is why a parent or a sibling may be the best donor in many situations—a genetic match between the MHC genes is much more likely and the organ is much less likely to be rejected.
Although matching all of the MHC genes can lower the risk for rejection, there are a number of additional gene products that also play a role in stimulating responses against grafted tissue. Because of this, no non-self grafted tissue is likely to completely avoid rejection. However, the more similar the MHC gene match, the more likely the graft is to be tolerated for a longer time. Most transplant recipients, even those with tissues well matched to their MHC genes, require treatment with immunosuppressant drugs for the rest of their lives. This can make them more vulnerable than the general population to complications from infectious diseases. It can also result in transplant-related malignancies because the body’s normal defenses against cancer cells are being suppressed.
Graft-versus-Host Disease
A form of rejection called graft-versus-host disease (GVHD) primarily occurs in recipients of bone marrow transplants and peripheral blood stem cells. GHVD presents a unique situation because the transplanted tissue is capable of producing immune cells; APCs in the donated bone marrow may recognize the host cells as non-self, leading to activation of the donor cytotoxic T cells. Once activated, the donor’s T cells attack the recipient cells, causing acute GVHD.
Acute GVHD typically develops within weeks after a bone marrow transplant, causing tissue damage affecting the skin, gastrointestinal tract, liver, and eyes. In addition, acute GVHD may also lead to a cytokine storm, an unregulated secretion of cytokines that may be fatal. In addition to acute GVHD, there is also the risk for chronic GVHD developing months after the bone marrow transplant. The mechanisms responsible for chronic GVHD are not well understood.
To minimize the risk of GVHD, it is critically important to match the HLAs of the host and donor as closely as possible in bone marrow transplants. In addition, the donated bone marrow is processed before grafting to remove as many donor APCs and T cells as possible, leaving mostly hematopoietic stem cells.
The Future of Transplantation
Historically speaking, the practice of transplanting tissues—and the complications that can accompany such procedures—is a relatively recent development. It was not until 1954 that the first successful organ transplantation between two humans was achieved. Yet the field of organ transplantation has progressed rapidly since that time.
Nonetheless, the practice of transplanting non-self tissues may soon become obsolete. Scientists are now attempting to develop methods by which new organs may be grown in vitro from an individual’s own harvested cells to replace damaged or abnormal ones. Because organs produced in this way would contain the individual’s own cells, they could be transplanted into the individual without risk for rejection.
An alternative approach that is gaining renewed research interest is genetic modification of donor animals, such as pigs, to provide transplantable organs that do not elicit an immune response in the recipient. The approach involves excising the genes in the pig (in the embryo) that are most responsible for the rejection reaction after transplantation. Finding these genes and effectively removing them is a challenge, however. So too is identifying and neutralizing risks from viral sequences that might be embedded in the pig genome, posing a risk for infection in the human recipient.
1.16 Immunodeficiency
Immunodeficiencies are inherited (primary) or acquired (secondary) disorders in which elements of host immune defenses are either absent or functionally defective. In developed countries, most immunodeficiencies are inherited, and they are usually first seen in clinics as recurrent or overwhelming infections in infants. However, on a global scale, malnutrition is the most common cause of immunodeficiency and would be categorized as an acquired immunodeficiency. Acquired immunodeficiencies are more likely to develop later in life, and the pathogenic mechanisms of many remain obscure.
Primary Immunodeficiency
Primary immunodeficiencies, which number more than 250, are caused by inherited defects of either nonspecific innate or specific adaptive immune defenses. In general, patients born with primary immunodeficiency (PI) commonly have an increased susceptibility to infection. This susceptibility can become apparent shortly after birth or in early childhood for some individuals, whereas other patients develop symptoms later in life. Some primary immunodeficiencies are due to a defect of a single cellular or humoral component of the immune system; others may result from defects of more than one component. Examples of primary immunodeficiencies include chronic granulomatous disease, X-linked agammaglobulinemia, selective IgA deficiency, and severe combined immunodeficiency disease.
Chronic Granulomatous Disease
The causes of chronic granulomatous disease (CGD) are defects in the NADPH oxidase system of phagocytic cells, including neutrophils and macrophages, that prevent the production of superoxide radicals in phagolysosomes. The inability to produce superoxide radicals impairs the antibacterial activity of phagocytes. As a result, infections in patients with CGD persist longer, leading to a chronic local inflammation called a granuloma. Microorganisms that are the most common causes of infections in patients with CGD include Aspergillus spp., Staphylococcus aureus, Chromobacterium violaceum, Serratia marcescens, and Salmonella typhimurium.
X-Linked Agammaglobulinemia
Deficiencies in B cells due to defective differentiation lead to a lack of specific antibody production known as X-linked agammaglobulinemia. In 1952, Ogden C. Bruton (1908–2003) described the first immunodeficiency in a boy whose immune system failed to produce antibodies. This defect is inherited on the X chromosome and is characterized by the absence of immunoglobulin in the serum; it is called Bruton X-linked agammaglobulinemia (XLA). The defective gene, BTK, in XLA is now known to encode a tyrosine kinase called Bruton tyrosine kinase (Btk). In patients whose B cells are unable to produce sufficient amounts of Btk, the B-cell maturation and differentiation halts at the pre-B-cell stage of growth. B-cell maturation and differentiation beyond the pre-B-cell stage of growth is required for immunoglobulin production. Patients who lack antibody production suffer from recurrent infections almost exclusively due to extracellular pathogens that cause pyogenic infections including: Haemophilus influenzae, Streptococcus pneumoniae, S. pyogenes, and S. aureus. Because cell-mediated immunity is not impaired, these patients are not particularly vulnerable to infections caused by viruses or intracellular pathogens.
Selective IgA Deficiency
The most common inherited form of immunoglobulin deficiency is selective IgA deficiency, affecting about one in 800 people. Individuals with selective IgA deficiency produce normal levels of IgG and IgM, but are not able to produce secretory IgA. IgA deficiency predisposes these individuals to lung and gastrointestinal infections for which secretory IgA is normally an important defense mechanism. Infections in the lungs and gastrointestinal tract can involve a variety of pathogens, including: H. influenzae, S. pneumoniae, Moraxella catarrhalis, S. aureus, Giardia lamblia, or pathogenic strains of Escherichia coli.
Severe Combined Immunodeficiency
Patients who suffer from severe combined immunodeficiency (SCID) have B-cell and T-cell defects that impair T-cell dependent antibody responses as well as cell-mediated immune responses. Patients with SCID also cannot develop immunological memory, so vaccines provide them no protection, and live attenuated vaccines (e.g., for varicella-zoster, measles virus, rotavirus, poliovirus) can actually cause the infection they are intended to prevent. The most common form is X-linked SCID, which accounts for nearly 50% of all cases and occurs primarily in males. Patients with SCID are typically diagnosed within the first few months of life after developing severe, often life-threatening, opportunistic infection by Candida spp., Pneumocystis jirovecii, or pathogenic strains of E. coli.

Without treatment, babies with SCID do not typically survive infancy. In some cases, a bone marrow transplant may successfully correct the defects in lymphocyte development that lead to the SCID phenotype, by replacing the defective component. However, this treatment approach is not without risks, as demonstrated by the famous case of David Vetter (1971–1984), better known as “Bubble Boy” (figure 1.58). Vetter, a patient with SCID who lived in a protective plastic bubble to prevent exposure to opportunistic microbes, received a bone marrow transplant from his sister. Because of a latent Epstein-Barr virus infection in her bone marrow, however, he developed mononucleosis and died of Burkitt lymphoma at the age of 12 years.
Secondary Immunodeficiency
A secondary immunodeficiency occurs as a result of an acquired impairment in the function of B cells, T cells, or both. Secondary immunodeficiencies can be caused by:
- Systemic disorders such as diabetes mellitus, malnutrition, hepatitis, or HIV infection
- Immunosuppressive treatments such as cytotoxic chemotherapy, bone marrow ablation before transplantation, or radiation therapy
- Prolonged critical illness due to infection, surgery, or trauma in the very young, elderly, or hospitalized patients
Unlike primary immunodeficiencies, which have a genetic basis, secondary immunodeficiencies are often reversible if the underlying cause is resolved. Patients with secondary immunodeficiencies develop an increased susceptibility to an otherwise benign infection by opportunistic pathogens such as Candida spp., P. jirovecii, and Cryptosporidium.
HIV infection and the associated acquired immunodeficiency syndrome (AIDS) are the best-known secondary immunodeficiencies. AIDS is characterized by profound CD4 T-cell lymphopenia (decrease in lymphocytes). The decrease in CD4 T cells is the result of various mechanisms, including HIV-induced pyroptosis (a type of apoptosis that stimulates an inflammatory response), viral cytopathic effect, and cytotoxicity to HIV-infected cells.
The most common cause of secondary immunodeficiency worldwide is severe malnutrition, which affects both innate and adaptive immunity. More research and information are needed for the more common causes of secondary immunodeficiency; however, the number of new discoveries in AIDS research far exceeds that of any other single cause of secondary immunodeficiency. AIDS research has paid off extremely well in terms of discoveries and treatments; increased research into the most common cause of immunodeficiency, malnutrition, would likely be as beneficial.
Table 1.22 summarizes primary and secondary immunodeficiencies, their effects on immune function, and typical outcomes.
| Disease | Effect on Immune Function | Outcomes |
|---|---|---|
| Primary immunodeficiencies | ||
| Chronic granulomatous disease | Impaired killing of bacteria within the phagolysosome of neutrophils and macrophages | Chronic infections and granulomas |
| Selective IgA deficiency | Inability to produce secretory IgA | Predisposition to lung and gastrointestinal infections |
| Severe combined immunodeficiency disease (SCID) | Deficient humoral and cell-mediated immune responses | Early development of severe and life-threatening opportunistic infections |
| X-linked agammaglobulinemia | Flawed differentiation of B cells and absence of specific antibodies | Recurrent infections almost exclusively due to pathogens that cause pyogenic infections |
| Secondary immunodeficiencies | ||
| Immunosuppressive therapies (e.g., chemotherapy, radiotherapy) | Impaired humoral and/or cell-mediated immune responses | Opportunistic infections, rare cancers |
| Malnutrition | Impaired humoral and/or cell-mediated immune responses | Opportunistic infections, rare cancers |
| Viral infection (e.g., HIV) | Impaired cell-mediated immune responses due to CD4 T-cell lymphopenia | Opportunistic infections, rare cancers |
Table 1.22: Primary and Secondary Immunodeficiencies
1.17 Cancer Immunobiology and Immunotherapy
Cancer involves a loss of the ability of cells to control their cell cycle, the stages each eukaryotic cell goes through as it grows and then divides. When this control is lost, the affected cells rapidly divide and often lose the ability to differentiate into the cell type appropriate for their location in the body. In addition, they lose contact inhibition and can start to grow on top of each other. This can result in formation of a tumor. It is important to make a distinction here: The term cancer is used to describe the diseases resulting from loss of cell-cycle regulation and subsequent cell proliferation. But the term tumor is more general. A tumor is an abnormal mass of cells, and a tumor can be benign (not cancerous) or malignant (cancerous).
Traditional cancer treatment uses radiation and/or chemotherapy to destroy cancer cells; however, these treatments can have unwanted side effects because they harm normal cells as well as cancer cells. Newer, promising therapies attempt to enlist the patient’s immune system to target cancer cells specifically. It is known that the immune system can recognize and destroy cancerous cells, and some researchers and immunologists also believe, based on the results of their experiments, that many cancers are eliminated by the body’s own defenses before they can become a health problem. This idea is not universally accepted by researchers, however, and needs further investigation for verification.
Cell-Mediated Response to Tumors
Cell-mediated immune responses can be directed against cancer cells, many of which do not have the normal complement of self-proteins, making them a target for elimination. Abnormal cancer cells may also present tumor antigens. These tumor antigens are not a part of the screening process used to eliminate lymphocytes during development; thus, even though they are self-antigens, they can stimulate and drive adaptive immune responses against abnormal cells.
Presentation of tumor antigens can stimulate naïve helper T cells to become activated by cytokines such as IL-12 and differentiate into TH1 cells. TH1 cells release cytokines that can activate natural killer (NK) cells and enhance the killing of activated cytotoxic T cells. Both NK cells and cytotoxic T cells can recognize and target cancer cells as well as induce apoptosis through the action of perforins and granzymes. In addition, activated cytotoxic T cells can bind to cell-surface proteins on abnormal cells and induce apoptosis by a second killing mechanism called the CD95 (Fas) cytotoxic pathway.
Despite these mechanisms for removing cancerous cells from the body, cancer remains a common cause of death. Unfortunately, malignant tumors tend to actively suppress the immune response in various ways. In some cancers, the immune cells themselves are cancerous. In leukemia, lymphocytes that would normally facilitate the immune response become abnormal. In other cancers, the cancerous cells can become resistant to induction of apoptosis. This may occur through the expression of membrane proteins that shut off cytotoxic T cells or that induce regulatory T cells that can shut down immune responses.
The mechanisms by which cancer cells alter immune responses are still not fully understood, and this is a very active area of research. As scientists’ understanding of adaptive immunity improves, cancer therapies that harness the body’s immune defenses may someday be more successful in treating and eliminating cancer.
Cancer Vaccines
There are two types of cancer vaccines: preventive and therapeutic. Preventive vaccines are used to prevent cancer from occurring, whereas therapeutic vaccines are used to treat patients with cancer. Most preventive cancer vaccines target viral infections that are known to lead to cancer. These include vaccines against human papillomavirus (HPV) and hepatitis B, which help prevent cervical and liver cancer, respectively.
Most therapeutic cancer vaccines are in the experimental stage. They exploit tumor-specific antigens to stimulate the immune system to selectively attack cancer cells. Specifically, they aim to enhance TH1 function and interaction with cytotoxic T cells, which, in turn, results in more effective attack on abnormal tumor cells. In some cases, researchers have used genetic engineering to develop antitumor vaccines in an approach similar to that used for DNA vaccines. The vaccine contains a recombinant plasmid with genes for tumor antigens; theoretically, the tumor gene would not induce new cancer because it is not functional, but it could trick the immune system into targeting the tumor gene product as a foreign invader.
The first FDA-approved therapeutic cancer vaccine was sipuleucel-T (Provenge), approved in 2010 to treat certain cases of prostate cancer.[15] This unconventional vaccine is custom-designed using the patient’s own cells. APCs are removed from the patient and cultured with a tumor-specific molecule; the cells are then returned to the patient. This approach appears to enhance the patient’s immune response against the cancer cells. Another therapeutic cancer vaccine (talimogene laherparepvec, also called T-VEC or Imlygic) was approved by the FDA in 2015 for treatment of melanoma, a form of skin cancer. This vaccine contains a virus that is injected into tumors, where it infects and lyses the tumor cells. The virus also induces a response in lesions or tumors besides those into which the vaccine is injected, indicating that it is stimulating a more general (as opposed to local) antitumor immune response in the patient.
Figure Descriptions
Figure 1.1: Tight junctions – two membranes connected with many spot welds in multiple lines. Desmosomes – two membranes with long strands weaving them together. Gap junctions – two membranes with a few spot welds each of which has a pore in the center.
Figure 1.2: A diagram of a section of skin. The bottom layer is the hypodermis and is mostly made up of large circular cells (fatty tissue). The next layer up, and the thickest layer is the dermis. At the bottom of the dermis are blood vessels, lymph vessels, and nerves, all of which run throughout the dermis. Sweat glands are coiled tubes that lead to the surface. Hair follicles are thick vase-shaped structures containing a hair; an oil gland is attached to the hair follicle. The top layer is the epidermis and is made of many layers of flat cells.
Figure 1.3: A spongy-looking surface with tufts of long hairs. Each hair is about 5 µm long; each tuft is about 10 µm in diameter.
Figure 1.4: Figure a is a diagram of a single goblet cell. The cell is tall and slightly hourglass shaped. The bottom of the cell is filled with a nucleus. The top shows the Golgi apparatus (folds of membranes), rough endoplasmic reticulum (folds of membranes with dots), secretory vesicles containing mucin (large bubbles), and microvilli (finger-like projections at the top). Figure b is a micrograph of two goblet cells within a row of epithelial cells. The epithelial cells are rectangular with a large nucleus visible. The goblet cells are thinner and have a clear (uncolored) top.
Figure 1.5: A diagram of a person. An arrow from the eye points to a larger image that shows eyelashes, the eyelid, and tear ducts. An arrow from the abdominal region shows a larger kidney are ureter. An arrow from the groin region shows a larger bladder, including ureter pelvic floor muscles, and urethra.
Figure 1.6: A micrograph and diagram both show a large hair follicle (a vase-shaped pocket) with a hair projecting out past the epidermis. On the side of the hair follicle is the sebaceous gland, which is a lumpy structure.
Figure 1.7: A diagram outlining the three complement pathways. At the top is the invading pathogen. Two antibodies bind to an antigen on the surface of the pathogen. C1 binds to the antigen-antibody complex. This is labeled the classic pathway. C1 causes C2 and C4 to be cut into two pieces. Parts of C2 and C4 bind together to form C3 convertase. The alternate pathway also leads to C3 convertase but does so directly. C3 convertase then cuts C3 in two and one of these binds to C3 convertase. The resulting enzyme is called C5 convertase. C5 convertase lyses C5 into two pieces. One of the C5 pieces joins other complement proteins (C6, C7, C8 and C9) to create a pore through the membrane of the invading cell. This pore kills the cell. Endogenous proteins on the host cell protect the host membrane from the complement proteins.
Figure 1.8: A cell with viruses inside it releases signals labeled interferons. The interferons travel to 3 different cells. The interferon signals neighboring uninfected cells to destroy RNA and reduce protein synthesis. The interferon signals neighboring infected cells to undergo apoptosis. The interferon also activates immune cells.
Figure 1.9: A flowchart showing progression of development for formed elements of blood. At the top is a multipotent hematopoietic stem cell (hemocytoblast). This cell divides and after division some of the new cells remain stem cells. Others go down one of two paths depending on the chemical signals received. One path begins with lymphoid stem cells which can either become natural killer cells (large granular lymphocytes) or small lymphocytes. The natural killer cell is a medium-large purple cell. Small lymphocytes can either become T lymphocytes or B lymphocytes. The T and B lymphocytes are medium-sized cells with a large nucleus. B lymphocytes become plasma cells, which are medium-sized cells with a large nucleus. The other option for the stem cell is to become a myeloid stem cell. Myeloid stem cells follow one of four paths. One path leads to megakaryocyte which leads to platelets. Platelets are small flecks. The second path leads to erythrocyte. Erythrocytes are small donut-shaped red cells. The third path leads to mast cells. The fourth path leads to basophil, neutrophil, eosinophil, or monocyte. Basophils are medium cells with many dark purple spots. Neutrophils are medium pink cells with a multilobed nucleus. Eosinophils are medium size cells with many pink spots. Monocytes lead to macrophages or dendritic cells. Macrophages are large irregularly shaped cells. Dendritic cells have longer tendons branching off of them.
Figure 1.10: Neutrophils have a multi-lobed nucleus. Eosinophils have a two-lobed nucleus and distinct pink spots when stained. Basophils have a two-lobed nucleus and distinct purple spots when stained. Each type of granulocyte is illustrated with a micrograph above it.
Figure 1.11: a) Mast cells in blood. Mast cells are large purple cells, red blood cells are small pink cells with a clear center. b) mast cell outside of blood.
Figure 1.12: NK cells have both inhibitory and activating receptors. Normal cells have signals on their MHC molecules that bind to the inhibitory receptors; so the NK cell does not kill them. Cells that are infected with virus have ligands that bind to the activating receptor; this causes the NK cell to kill them.
Figure 1.13: Many red blood cells with a single larger cell. The larger cell is pink with a purple region that fills nearly the entire cell. The purple region is labeled perforin-containing granules.
Figure 1.14: Monocytes are large cells with a large purple nucleus. There is a cluster of them in a field of smaller red blood cells. A PMN is also visible with a dark, multi-lobed nucleus. Macrophages are large cells with a defined nucleus.
Figure 1.15: A diagram with 3 steps. The first step states: leukocytes in the blood respond to chemical attractants released by pathogens and chemical signals from nearby injured cells. An injury to the surface of the skin is labeled: injured/infected cells secrete chemical signals into the blood. Pathogens are present in the wound. Neutrophils and monocytes are in the bloodstream, and the outside of the vessel is labeled capillary epithelial cells. A resident macrophage engulfs the pathogens and releases proinflammatory chemotactic cytokines. The second step states: the leukocytes squeeze between the cells of the capillary wall as they follow the chemical signals to where they are most concentrated (positive chemotaxis). The leukocytes emigrate to the site of injury and infection. The chemical signals present include C5a and cytokines. The third panel states: Within the damaged tissue, neutrophils release chemicals that break apart pathogens. Monocytes differentiate into macrophages. Neutrophils and macrophages phagocytize pathogens and cellular debris. Neutrophils release cytotoxic chemicals from granules into tissue.
Figure 1.16: A cell with three receptors. The lipopeptide receptor binds lipopeptides, the flagellin receptor binds flagellin, and the peptidoglycan receptor binds peptidoglycans. A fourth receptor (the nucleic acid receptor which binds to nucleic acids) is found on the membrane of the phagosome. All four receptors have an arrow pointing to the nucleus which contains DNA. An arrow pointing out reads:production and secretion of antiviral interferons and other cytokines.
Figure 1.17: Pseudopods of the larger cell engulf a smaller cell labeled infectious bacterium. The resulting vesicle containing the bacterium is labeled phagosome. This fuses with a lysosome which contains digestive enzymes. The resulting vesicle is labeled phagolysosome. Exocytosis removes the remaining debris.
Figure 1.18: a) a diagram of a wound in the skin that has let pathogens enter. Mast cells release histamines, which signal to cells in the bloodstream. B) The cells have left the bloodstream; these phagocytes are engulfing the pathogens.
Figure 1.19: A micrograph of a tubercle which consists of many darkly staining cells that form a circular structure.
Figure 1.20: A diagram with exogenous pyrogen at the top. These activate leukocytes which in turn release IL-6. The leukocytes also produce pyrogenic cytokines (IL-1, TNF-α, IFN-γ) which lead to the production of IL-6. IL-6 signals the circumventricular organs of the brain to produce PGE2 which results in fever. The temperature-dependent feedback on cytokine expression decreases the production of IL-6 in a negative feedback loop.
Figure 1.21: A graph with time on the X axis and concentration of antibody on the Y axis. The concentration is near 0 at the initial exposure and increases during the primary immune response. The concentration then drops back down but remains above the level at initial exposure. The secondary exposure increases the concentration of antibody to higher levels than the primary response. And even after dropping back down this count remains relatively high.
Figure 1.22: A drawing of an antigen as a large sphere with different shapes on the surface labeled epitopes.
Figure 1.23: Many antigens (shown as large spheres) each with multiple shapes on the surface labeled epitopes. Different antibodies are shown each with a binding site specific to one of the epitopes.
Figure 1.24: a) An antibody is a Y shape made of four strands. The two inner strands form the actual Y shape and are the heavy chains. The two light chains sit on the outsides of the top regions of the Y. The bottom portion of the Y (made of only heavy chains) is called the Fc Region. The Fc region along with half of the top portion of the Y (made of both light and heavy chains) is the constant regions. The variable region is the very tips of the Y and is made of both light and heavy chains. The antigen binding site is in the variable region. Disulfide bridges hold the antigen’s shape. B) a space filling model of the antigen.
Figure 1.25: A virus is drawn as a circle with knobs on it. Antigens bind to the knobs, thereby surrounding the virus. Next image shows antibody binding to diphtheria toxin. Next image shows antibody binding to a bacterial cell.
Figure 1.26: A macrophage with projections that are engulfing a pathogen with antibodies attached to it.
Figure 1.27: Bacterial cells with various epitopes (shown as different shapes). IgM antibodies are bound to multiple bacteria; all attached to the same shaped epitope which matches their binding sites.
Figure 1.28: Fc receptors on an NK cell bind to the Fc region of the IgG bound to the antigen on the surface of a pathogen. This causes the NK cell to release toxins that kill the pathogen.
Figure 1.29: Drawing of a phospholipid bilayer (plasma membrane). An MHC Class I protein molecule is found in all nucleated body cells. It has a linear portion in the membrane and four portions on the outer side of the cell. One of these portions connects to the membrane-spanning portion; two form the antigen binding site; and the fourth is labeled the Beta-2 microglobulin. MHC Class II protein molecules are found in lymphocytes and macrophages. This has two membrane-spanning portions (each attached to a portion on the outside of the cell). The two portions attached to these form the antigen-binding site.
Figure 1.30: The process of phagocytosis. 1: A bacterium is engulfed by phagocytosis into a dendritic cell and is encased in a phagosome. 2: Lysosomes fuse with the phagosome and digest the bacterium. 3: Immunodominant epitopes are associated with MHC II and presented on the cell surface.
Figure 1.31: A micrograph of a round cell approximately 7 micrometers in diameter. The cell has a studded surface.
Figure 1.32: a) A drawing of a femur; a long bone with a round head. B) A cross-section of the head of the femur.
Figure 1.33: A drawing of the thymus (a structure sitting on the surface of the heart); the spleen (a kidney-shaped structure in the upper left abdomen; the right lymphatic duct entering vein (a tube in the neck); lymph nodes (enlarged regions of lymph ducts); and a tonsil in the cheek. A callout shows a micrograph of the thymus which has a surface layer labeled fibrous capsule, a central tissue labeled medulla, out tissue labeled cortex, and lighter branches in the cortex labeled trabeculae.
Figure 1.34: Drawing of two bars spanning the T cell plasma membrane. On one side of the membrane is the intracellular domain. The transmembrane region spans the membrane. The constant region is outside the membrane; a disulfide bond holds these two bars together in the constant region. The variable region is at the top and contains the antigen-binding sites.
Figure 1.35: A native helper T cell binds to an antigen extracted from a pathogen sitting on the MCH Class II protein of an antigen-presenting cell (dendritic cell). The portion of the helper T cell that binds is the T cell receptor and is stabilized by CD4. After this binding the helper T cell is activated and can become TH1 cell; TH2 cell or memory helper T cell.
Figure 1.36: A naïve Cytotoxid T cell has a T cell receptor and CD8. The T cell receptor binds to an antigen extracted from a pathogen on the MHC class I of an infected pathogen. The CD8 stabilizes this reaction. This causes the activated CTL to release granzymes and perforins, which result in the controlled destruction of the infected cell through apoptosis.
Figure 1.37: a) the T cell receptor on the T cell recognizes the epitope on the MHC II on the macrophage and binds. B) The T cell receptor binds even though it does not recognize the epitope because the superantigen is bound. Many dots labeled cytokines are present.
Figure 1.38: A B cell plasma membrane has two long rectangles spanning it; these form a Y shape. Two shorter rectangles sit on the outside of the upper portion of the Y. The region spanning the membrane and halfway through the bars of the Y is the constant region. The upper region is the variable region which has the antigen binding sites. The long rectangles are the heavy chains. The shorter rectangles are the light chains. Multiple disulfide bridges hold the constant region together.
Figure 1.39: A circle with small chains of hexagons projecting from the surface is a pathogenic bacterial cell. The chains are polysaccharide antigens with repeating epitopes. Antibodies on the B cell bind to these epitopes. This causes the activation of the B cell and secretion of pentameric IgM.
Figure 1.40: 1: BCR interaction with antigen on intact pathogen. An antigen on the surface of a bacterium binds to the B cell receptor on the B cell. s: Antigen processing and presentation with MHC II. The antigen is on the MHC II. 3: Antigen presentation and activation of helper T cell. T cell receptor of helper T cell binds to antigen on MHCII. This is stabilized by CD4. Helper T releases cytokines. 4: Cytokines stimulate clonal proliferation and differentiation into memory B cells and antibody-secreting plasma cells.
Figure 1.41: A graph with time on the X-axis and antibody concentration in serum. At first there is very little antibody (near 0). The lag period does not see a significant increase. In the primary response, IgM peaks for about 5 days and drops. At the same time IgG increases and then drops. This creates an increase in antibody count with a plateau of about 5 days as both antibody types are present. The secondary response sees a prolonged peak of IgG with a peak of IgM for about 1 to 2 days at about the same time as the peak of IgG. The total antibody is also higher but isn’t at its plateau for as long as it is in the primary response.
Figure 1.42: a) Painting of Edward Jenner. B) Photo of many red lumps on the skin.
Figure 1.43: A) micrograph of pollen granules in different shapes and with different surface features. B) photo of a rash on a person’s back. C) Photo of peanuts.
Figure 1.44: Drawing of TH2 cell response. 1: Upon first exposure to allergen, antigen-presenting cell processes antigen and presents it to TH2 Cell. A large antigen-presenting cell is shown engulfing an antigen which is attached to a Class II MHC inside the cell. This class II MHC is then placed on the surface with the antigen on the end of the MHC. The TH2 cell has a receptor that binds to the antigen on the MHC. 2: TH2 cell releases IL-4 and IL-13 which activates B cell. The TH2 cell has unbound from the antigen-presenting cell and binds to a B cell with the antigen on it’s MHC and antibodies. The TH2 cell then releases small dots. 3: B cells proliferate and differentiate into plasma cells that synthesize and secrete IgE antibody. B cell is shown dividing. These cells then become plasma cells which are larger and are producing many IgE 4: IgE binds to mast cells by Fc region, sensitizing the mast cells. Mast cell is shown with IgE bound to it. 5: Upon subsequent exposure to allergen, mast cells with IgE bind to antigen and release inflammatory molecules, resulting in allergy symptoms. Antigen is shown bound to mast cell and the mast cell is releasing little dots labeled inflammatory molecules.
Figure 1.45: Table of Blood Types. Type A blood has red blood cells with A antigens as surface markers. It produces anti-B isohemagglutinins. Type B blood has red blood cells with B antigens as surface markers. It produces anti-A isohemagglutinins. Type AB blood has red blood cells with both A and B antigens as surface markers. It produces neither isohemagglutinins. Type O blood has red blood cells with neither A nor B antigens as surface markers. It produces both anti-A and anti-B isohemagglutinins.
Figure 1.46: Diagram of a Type A donor giving blood to a Type B recipient. 1: Donated type A blood with type A antigens enters bloodstream of type B recipient. Red blood cells with Type A antigens and red blood cells with Type B antigens are shown; anti-A antibody is also present. 2: Anti-A antibodies in plasma of Type B recipient bind to donated type A red blood cells. 3: Bound anti-A antibodies activate complement cascade, releasing hemoglobulin and destroying red blood cells. Small dots are shown destroying type A cells.
Figure 1.47: First pregnancy with Rh+ fetus resulting in healthy newborn. The diagram shows an Rh- person and an Rh+ fetus. Rh+ red blood cells cross the placenta into pregnant person’s circulation. This causes anti-Rh antibodies to be produced in the person upon exposure to fetal Rh antigens. The second pregnancy with Rh+ fetus results in hemolytic newborn. The diagram shows an Rh- person with an Rh+ fetus. Anti-Rh antibodies remain in pregnant person’s circulation from the first pregnancy and cross the placenta. Maternal anti-Rh antibodies attack and destroy fetal Rh+ red blood cells. b) First pregnancy with Rh+ fetus and anti-Rh antibody treatment resulting in healthy newborn The diagram shows an Rh- person and an Rh+ fetus. Rh+ red blood cells from the fetus cross placenta into pregnant person’s circulation. Anti-Rh antibodies (Rhogam) bind and inactivate fetal Rh antigens before they stimulate immune response in the pregnant person.
Figure 1.48: IgG binds to antigens and forms an immune complex made of multiple IgG and antigens. Immune complex deposition in tissues, activation of complement and inflammation. Image shows Immune complex binding to tissues, binding to C1, C3b and C5a binding to tissues. And IgG binding to neutrophils. Tissue damage is shown. B) Person on a dialysis machine.
Figure 1.49: a) Sensitization. Antigens from poison ivy enter dendritic cells in skin. The dendritic cell activates a T-cell which becomes a memory helper T cell. b) Immune response. Macrophages, memory helper T cells, and cytotoxic T cells produce a large lesion on the skin due to cytokine activation.
Figure 1.50: A) Photo of chickens in a coop. b) Photo of a worker in a warehouse.
Figure 1.51: Photo of a person with many dots in a row on their skin. The dots are numbered and marks are next to those that are swollen.
Figure 1.52: A person with a large swollen neck.
Figure 1.53: Photo of a person with large bulging eyes.
Figure 1.54: Two hands, palm up are shown. They appear to be more red than is normal.
Figure 1.55: a) Diagram of a normal nerve cell releasing acetylcholine which binds to receptors on the muscle cell. This signal is processed and the muscle cell contracts. B) Diagram of myasthenia gravis. The nerve cell releases acetylcholine but anti-AChR antibodies bind to the acetylcholine so it cannot bind to the receptors on the muscle cells. Because the signal is blocked the muscle is paralyzed and does not contract.
Figure 1.56: X-ray and photo of hands with joints bent at unusual angles.
Figure 1.57: a) Diagram of symptoms include: a rash on the phase, ulcers of the nose and mouth, muscle aches, inflammation of the pericardium (heart region), poor circulation in the fingers and toes. B) photo of a butterfly rash on the face.
Figure 1.58: Photo of a boy in a suit similar to a space suit.
Figure References
Figure 1.1: There are multiple types of cell junctions in human tissue, three of which are shown here. Modification of work by (c) Mariana Ruiz Villareal. CC BY 4.0.
Figure 1.2: Human skin has three layers: the epidermis, the dermis, and the hypodermis. Modification of work by National Institutes of Health. Public Domain. https://commons.wikimedia.org/wiki/File:Anatomy_The_Skin_-_NCI_Visuals_Online.jpg
Figure 1.3: This scanning electron micrograph shows ciliated and nonciliated epithelial cells from the human trachea. Charles Daghlian. Public Domain. https://commons.wikimedia.org/wiki/File:Bronchiolar_epithelium_3_-_SEM.jpg
Figure 1.4: Goblet cells produce and secrete mucus. (c) 2012. Regents of University of Michigan Medical School. Redistribution authorized with attribution.
Figure 1.5: Tears flush microbes away from the surface of the eye. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.6: Sebaceous glands secrete sebum, a chemical mediator that lubricates and protects the skin from invading microbes. (c) 2012. Regents of University of Michigan Medical School. Redistribution authorized with attribution.
Figure 1.7: The three complement activation pathways have different triggers, as shown here, but all three result in the activation of the complement protein C3, which produces C3a and C3b. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Table 1.5: Autocrine, paracrine, and endocrine actions describe which cells are targeted by cytokines and how far the cytokines must travel to bind to their intended target cells’ receptors. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.8: Interferons are cytokines released by a cell infected with a virus. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.9: All the formed elements of the blood arise by differentiation of hematopoietic stem cells in the bone marrow. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.10: Granulocytes can be distinguished by the number of lobes in their nuclei and the staining properties of their granules. Top Left: “neutrophil” micrograph: modification of work by (c) Ed Uthman. CC BY 4.0. Top Middle and Right: Public Domain. Bottom: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.11: Mast cells function similarly to basophils by inducing and promoting inflammatory responses. (a) Credit: Left: Copyright unknown. (c) Right: Modification of Figure 1C. in Greenland, J.R., Xu, X., Sayah, D.M. et al. Mast cells in a murine lung ischemia-reperfusion model of primary graft dysfunction. Respir Res 15, 95 (2014). https://doi.org/10.1186/s12931-014-0095-0 CC BY 4.0.
Figure 1.12: Natural killer (NK) cells are inhibited by the presence of the major histocompatibility cell (MHC) receptor on healthy cells. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.13: Natural killer cell with perforin-containing granules. Modification of Figure 1 in Rolstad B (2014) The early days of NK cells: an example of how a phenomenon led to detection of a novel immune receptor system – lessons from a rat model. Front. Immunol. 5:283. https://doi.org/10.3389/fimmu.2014.00283 CC BY 4.0
Figure 1.14: Monocytes are large, agranular white blood cells with a nucleus that lacks lobes. Left: Modification of work by Armed Forces Institute of Pathology. Public Domain. https://commons.wikimedia.org/wiki/File:AML-M4.jpg. Right: Modification of work by Centers for Disease Control and Prevention/Martin D. Hicklin. Public Domain. https://phil.cdc.gov/Details.aspx?pid=6625
Figure 1.15: Damaged cells and macrophages that have ingested pathogens release cytokines that are proinflammatory and chemotactic for leukocytes. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.16: Phagocytic cells contain pattern recognition receptors (PRRs) capable of recognizing various pathogen-associated molecular patterns (PAMPs). (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.17: The stages of phagocytosis include the engulfment of a pathogen, the formation of a phagosome, the digestion of the pathogenic particle in the phagolysosome, and the expulsion of undigested materials from the cell. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.18: Mast cells detect injury to nearby cells and release histamine, initiating an inflammatory response. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.19: A tubercle is a granuloma in the lung tissue of a patient with tuberculosis. Modified figure by 1C in (c) Piotrowski WJ, Górski P, Duda-Szymańska J, Kwiatkowska S. Mycobacterium tuberculosis as a sarcoid factor? A case report of family sarcoidosis. Am J Case Rep. 2014 May 16;15:216-20. CC BY-NC-ND 3.0 Unported. https://pmc.ncbi.nlm.nih.gov/articles/PMC4026149. Reproduced under Fair Use.
Figure 1.20: The role of the hypothalamus in the inflammatory response. Left: Public Domain. https://commons.wikimedia.org/wiki/File:Hypothalamus.jpg. Right: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.21: This graph illustrates the primary and secondary immune responses related to antibody production after an initial and secondary exposure to an antigen. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.22: An antigen is a macromolecule that reacts with components of the immune system. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.23: A typical protein antigen has multiple epitopes, shown by the ability of three different antibodies to bind to different epitopes of the same antigen. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.24: The typical four-chain structure of a generic antibody monomer. Credit a: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology. Credit b: modification of work by Tim Vickers. Public Domain. https://commons.wikimedia.org/wiki/File:Antibody_IgG2.png
Table 1.9: The five immunoglobulin (Ig) classes. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.25: Neutralization involves the binding of specific antibodies to antigens found on bacteria, viruses, and toxins, preventing them from attaching to target cells. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.26: Antibodies serve as opsonins and inhibit infection by tagging pathogens for destruction by macrophages, dendritic cells, and neutrophils. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.27: Antibodies, especially IgM antibodies, agglutinate bacteria by binding to epitopes on two or more bacteria simultaneously. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.28: In this example of ADCC, antibodies bind to a large pathogenic cell that is too big for phagocytosis and then bind to Fc receptors on the membrane of a natural killer cell. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.29: MHC I are found on all nucleated body cells, and MHC II are found on macrophages, dendritic cells, and B cells (along with MHC I). (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.30: A dendritic cell phagocytoses a bacterial cell and brings it into a phagosome. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.31: This scanning electron micrograph shows a T lymphocyte, which is responsible for the cell-mediated immune response. Modification of work by National Cancer Institute. Public Domain. https://commons.wikimedia.org/wiki/File:Red_White_Blood_cells.jpg
Figure 1.32: (a) Red bone marrow can be found in the head of the femur (thighbone) and is also present in the flat bones of the body, such as the ilium and the scapula. Left: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology. Right: Stevenfruitsmaak. Public Domain. https://commons.wikimedia.org/wiki/File:Caput_femoris_cortex_medulla.jpg
Figure 1.33: The thymus is a bi-lobed, H-shaped glandular organ that is located just above the heart. Left: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology. Right: (c) 2012. Regents of University of Michigan Medical School. Redistribution authorized with attribution.
Figure 1.34: A T-cell receptor spans the cytoplasmic membrane and projects variable binding regions into the extracellular space to bind processed antigens associated with MHC I or MHC II molecules. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.35: This illustration depicts the activation of a naïve (unactivated) helper T cell by an antigen-presenting cell and the subsequent proliferation and differentiation of the activated T cell into different subtypes. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.36: This figure illustrates the activation of a naïve (unactivated) cytotoxic T cell (CTL) by an antigen-presenting MHC I molecule on an infected body cell. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.37: (a) The macrophage in this figure is presenting a foreign epitope that does not match the TCR of the T cell. Because the T cell does not recognize the epitope, it is not activated. Modification of work by (c) “Microbiotic”/YouTube. CC BY 4.0.
Figure 1.38: B-cell receptors are embedded in the membranes of B cells. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.39: T-independent antigens have repeating epitopes that can induce B cell recognition and activation without involvement from T cells. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.40: In T cell-dependent activation of B cells, the B cell recognizes and internalizes an antigen and presents it to a helper T cell that is specific to the same antigen. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.41: Compared to the primary response, the secondary antibody response occurs more quickly and produces antibody levels that are higher and more sustained. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Table 1.12: The four mechanisms of acquisition of immunity. Top left: by USDA/Ken Hammond. Public Domain. https://commons.wikimedia.org/wiki/File:Breastfeeding_infant.jpg; credit top right photo: by Michaelberry https://commons.wikimedia.org/wiki/File:Intravenous_therapy_2007-SEP-13-Singapore.JPG; bottom left photo: Centers for Disease Control and Prevention. Public Domain. https://commons.wikimedia.org/wiki/File:Varicella.jpg; credit bottom right photo: Airman 1st Class Destinee Doughert / U.S. Air Force. Public Domain. https://nara.getarchive.net/media/us-air-force-senior-airman-tamika-bradley-20th-medical-feec7e
Figure 1.42: A painting of Edward Jenner depicts a cow and a milkmaid in the background. Public Domain. https://commons.wikimedia.org/wiki/File:Edward_Jenner.jpg; Right: Figure 1 from Palkonen P. et al. 2003. “Cowpox with Severe Generalized Eruption, Finland” Emerging Infections Diseases. 9 (11): 1458-1461. https://wwwnc.cdc.gov/eid/past-issues/volume-9. Public Domain.
Figure 1.43: a. Allergens in plant pollen. Dartmouth Electron Microscope Facility. Public Domain. https://commons.wikimedia.org/wiki/File:Misc_pollen_colorized.jpg. b. Hives. Modification of work of DLdoubleE. Public Domain. https://commons.wikimedia.org/wiki/File:Hives_on_back.jpg. c. Peanuts. Public Domain.
Figure 1.44: On first exposure to an allergen in a susceptible individual, antigen-presenting cells process and present allergen epitopes with major histocompatibility complex (MHC) II to T helper cells. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.45: Blood types. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.46: A type II hypersensitivity hemolytic transfusion reaction (HTR) leading to hemolytic anemia. Blood from a type A donor is administered to a patient with type B blood. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.47: When an Rh− mother has an Rh+ fetus, fetal erythrocytes are introduced into the mother’s circulatory system before or during birth, leading to production of anti-Rh IgG antibodies. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.48: Type III hypersensitivities and the systems they affect. Left: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology. Right: Tech. Sgt. Bennie J. Davis III / US Air Force; Public Domain. https://www.af.mil/News/Photos/igphoto/2000316503
Figure 1.49: Exposure to hapten antigens in poison ivy can cause contact dermatitis, a type IV hypersensitivity. Illustrations: (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology. Left: Adapted under Fair Use http://blogimages.serenataassets.com/pollennation/wp-content/uploads/2014/09/poison-ivy-160×160.jpg. Right: modification of work by Larsonja. Public Domain. https://commons.wikimedia.org/wiki/File:Urushiol_induced_contact_dermatitis.jpg
Figure 1.50: Occupational exposure to dust, mold, and other allergens can result in hypersensitivity pneumonitis. Left: Modification of work (c) The Global Orphan Project. CC BY 4.0. Right: kytrangho. Public Domain. https://pixabay.com/photos/gruyere-cheese-factory-switzerland-272462
Figure 1.51: Results of an allergy skin-prick test to test for type I hypersensitivity to a group of potential allergens. Modification of work (c) OakleyOriginals. CC BY 4.0. https://flic.kr/p/wx3wWU
Figure 1.52: Goiter, a hypertrophy of the thyroid, is a symptom of Graves disease and Hashimoto thyroiditis. By Almazi. Public Domain. https://commons.wikimedia.org/wiki/File:Goitre.jpg
Figure 1.53: Exophthalmia, or Graves ophthalmopathy, is a sign of Graves disease. Modification of work (c) Jonathan Trobe, University of Michigan Kellogg Eye Center. CC BY 3.0 Unported. https://commons.wikimedia.org/wiki/File:Proptosis_and_lid_retraction_from_Graves%27_Disease.jpg
Figure 1.54: Hyperpigmentation is a sign of Addison disease. Modification of work (c) Petros Perros. CC BY 2.5 Generic. https://commons.wikimedia.org/wiki/File:A_69-Year-Old_Female_with_Tiredness_and_a_Persistent_Tan_02.png
Figure 1.55: Myasthenia gravis and impaired muscle contraction. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 1.56: The radiograph (left) and photograph (right) show damage to the hands typical of rheumatoid arthritis. Left: Jojo. Public Domain. https://commons.wikimedia.org/wiki/File:Reumatoidalne_zapalenie_stawow_01.jpg. Right: (c) modification of work (c) handarmdoc. CC BY 2.0. https://flic.kr/p/f35DuE
Figure 1.57: Systemic lupus erythematosus. Left: Modification of work by Mikael Häggström. https://commons.wikimedia.org/wiki/File:Symptoms_of_SLE.svg. Public Domain. Right: Figure 4 from Shrestha D, Dhakal AK, Shiva RK, Shakya A, Shah SC, Shakya H. Systemic lupus erythematosus and granulomatous lymphadenopathy. BMC Pediatr. 2013 Nov 5;13:179. doi: 10.1186/1471-2431-13-179. CC BY 2.0.
Figure 1.58: David Vetter, popularly known as “The Bubble Boy,” was born with SCID and lived most of his life isolated inside a plastic bubble. NASA Johnson Space Center. Public domain.
Text References
- N. Parrow et al. “Sequestration and Scavenging of Iron in Infection.” Infection and Immunity 81 no. 10 (2013):3503–3514. ↵
- Blaschitz C., Raffatellu M. “Th17 cytokines and the gut mucosal barrier.” J Clin Immunol. 2010 Mar; 30(2):196-203. doi: 10.1007/s10875-010-9368-7. ↵
- K. Mupapa, M. Massamba, K. Kibadi, K. Kivula, A. Bwaka, M. Kipasa, R. Colebunders, J. J. Muyembe-Tamfum. “Treatment of Ebola Hemorrhagic Fever with Blood Transfusions from Convalescent Patients.” Journal of Infectious Diseases 179 Suppl. (1999): S18–S23. ↵
- N. J. Willis. “Edward Jenner and the Eradication of Smallpox.” Scottish Medical Journal 42 (1997): 118–121. ↵
- D.S. Strayer et al (eds). Rubin’s Pathology: Clinicopathologic Foundations of Medicine. 7th ed. 2, Philadelphia, PA: Lippincott, Williams & Wilkins, 2014. ↵
- C.M. Fitzsimmons et al. “Helminth Allergens, Parasite-Specific IgE, and Its Protective Role in Human Immunity.” Frontier in Immunology 5 (2015):47. ↵
- E.C. Vamvakas, M.A. Blajchman. “Transfusion-Related Mortality: The Ongoing Risks of Allogeneic Blood Transfusion and the Available Strategies for Their Prevention.” Blood 113 no. 15 (2009):3406–3417. ↵
- Reali, G., “Forty Years of Anti-D Immunoprophylaxis.” Blood Transfusion 5 no. 1 (2007):3–6. ↵
- C.L. Schneider et al. “A Pilot Study of Omalizumab to Facilitate Rapid Oral Desensitization in High-Risk Peanut-Allergic Patients.” Journal of Allergy and Clinical Immunology 132 no. 6 (2013):1368–1374. ↵
- P. Varshney et al. “A Randomized Controlled Study of Peanut Oral Immunotherapy: Clinical Desensitization and Modulation of the Allergic Response.” Journal of Allergy and Clinical Immunology 127 no. 3 (2011):654–660. ↵
- D.A. Van Heel, J. West. “Recent Advances in Coeliac Disease.” Gut 55 no. 7 (2006):1037—1046. ↵
- ibid. ↵
- P. Martorell et al. “Autoimmunity in Addison’s Disease.” Netherlands Journal of Medicine 60 no. 7 (2002):269—275. ↵
- C.C. Mok, C.S. Lau. “Pathogenesis of Systemic Lupus Erythematosus.” Journal of Clinical Pathology 56 no. 7 (2003):481—490. ↵
- National Institutes of Health, National Cancer Institute. "Cancer Vaccines." http://www.cancer.gov/about-cancer/causes-prevention/vaccines-fact-sheet#q8. Accessed on May 20, 2016. ↵

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}