2 Neurotransmitters — ACh, glutamate, GABA, and glycine
Learning Objectives
- Define the characteristics of a classical neurotransmitter.
- Describe the synthetic pathways, inactivation mechanisms, and neurochemical anatomy and mechanisms of receptor transduction for the following classical and nonclassical neurotransmitters:
- Acetylcholine
- Glutamate
- GABA (γ-aminobutyric acid)
- Glycine
- Catecholamines: dopamine, norepinephrine, epinephrine
- Histamine
- Serotonin (5-hydroxytryptaine)
- Review the major receptor classifications and representative receptor agonists and antagonists for the above transmitters.
- Compare steps of Gs protein activation versus steps that lead to an increase in intracellular Ca2+ and activation of protein kinase C following activation of a Gq protein.
Neurotransmitters are classically defined as:
- Being located in the neuron and produced by the neuron;
- Being released into the synaptic cleft when the neuron is stimulated;
- Acting on a postsynaptic receptor when released and causing a biological effect; and
- Being inactivated after they are released.
Acetylcholine
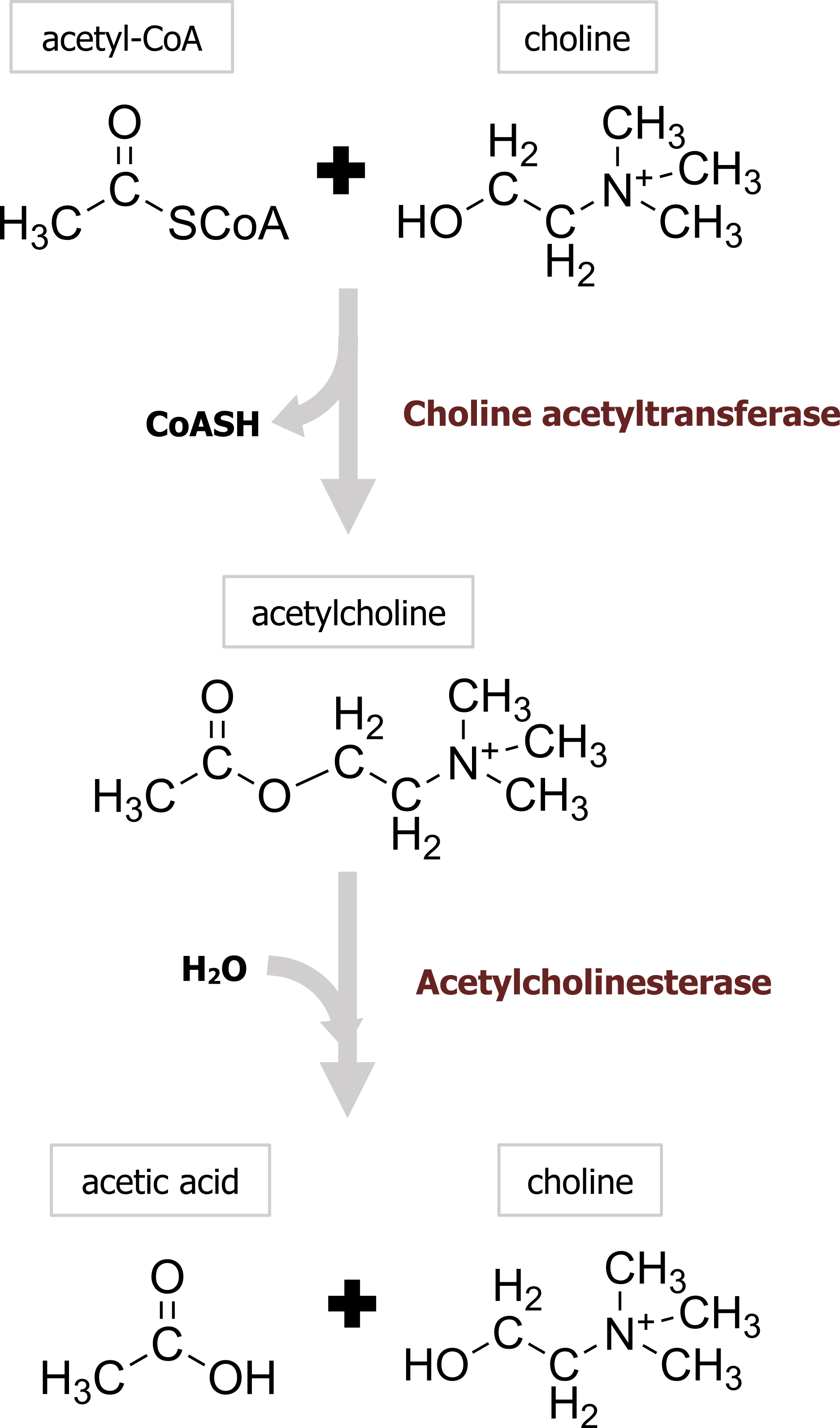
Acetylcholine (ACh) was the first identified neurotransmitter and is synthesized in nerve terminals from the precursors acetyl-CoA and choline, in a reaction catalyzed by choline acetyltransferase (ChAT) (figure 2.1). After synthesis in the cytoplasm of the neuron, a vesicular ACh transporter (VAChT) loads ACh into each cholinergic vesicle. The energy required to concentrate ACh within the vesicle is provided by the acidic pH of the vesicle lumen, which allows the VAChT to exchange H+ for ACh.
Termination of signal
In contrast to most other small-molecule neurotransmitters, the postsynaptic actions of ACh are not terminated by reuptake but hydrolysis by acetylcholinesterase (AChE). This enzyme is concentrated in the synaptic cleft, ensuring a rapid decrease in ACh concentration after its release from the presynaptic terminal. The hydrolysis results in acetate and choline (figure 2.1), which is recycled by being transported back into nerve terminals, where it is used to resynthesize ACh. Organophosphates are one class of drugs known to interact with ACh signal transmission through the inhibition of AChE, allowing ACh to accumulate at cholinergic synapses. This buildup of ACh depolarizes the postsynaptic muscle cell and renders it refractory to subsequent ACh release, causing neuromuscular paralysis.
Acetylcholine receptors
Many of the postsynaptic actions of ACh are mediated by the nicotinic ACh receptor (nAChR). nAChRs are nonselective cation channels that generate excitatory postsynaptic responses. Nicotinic receptors are large protein complexes consisting of five subunits. At the neuromuscular junction, the nAChR contains two α subunits, each of which has a binding site that binds a single molecule of ACh. Both ACh binding sites must be occupied for the receptor to be activated. In summary, the nAChR is a ligand-gated ion channel.
A second class of ACh receptors are referred to as muscarinic ACh receptors (mAChRs). mAChRs are metabotropic and mediate most of the effects of ACh in the brain. Like other metabotropic receptors, mAChRs have seven helical membrane-spanning domains. Binding of ACh to the receptor causes a conformational change that permits G-proteins to bind to the cytoplasmic domain of the mAChR (figure 2.2).

Glutamate
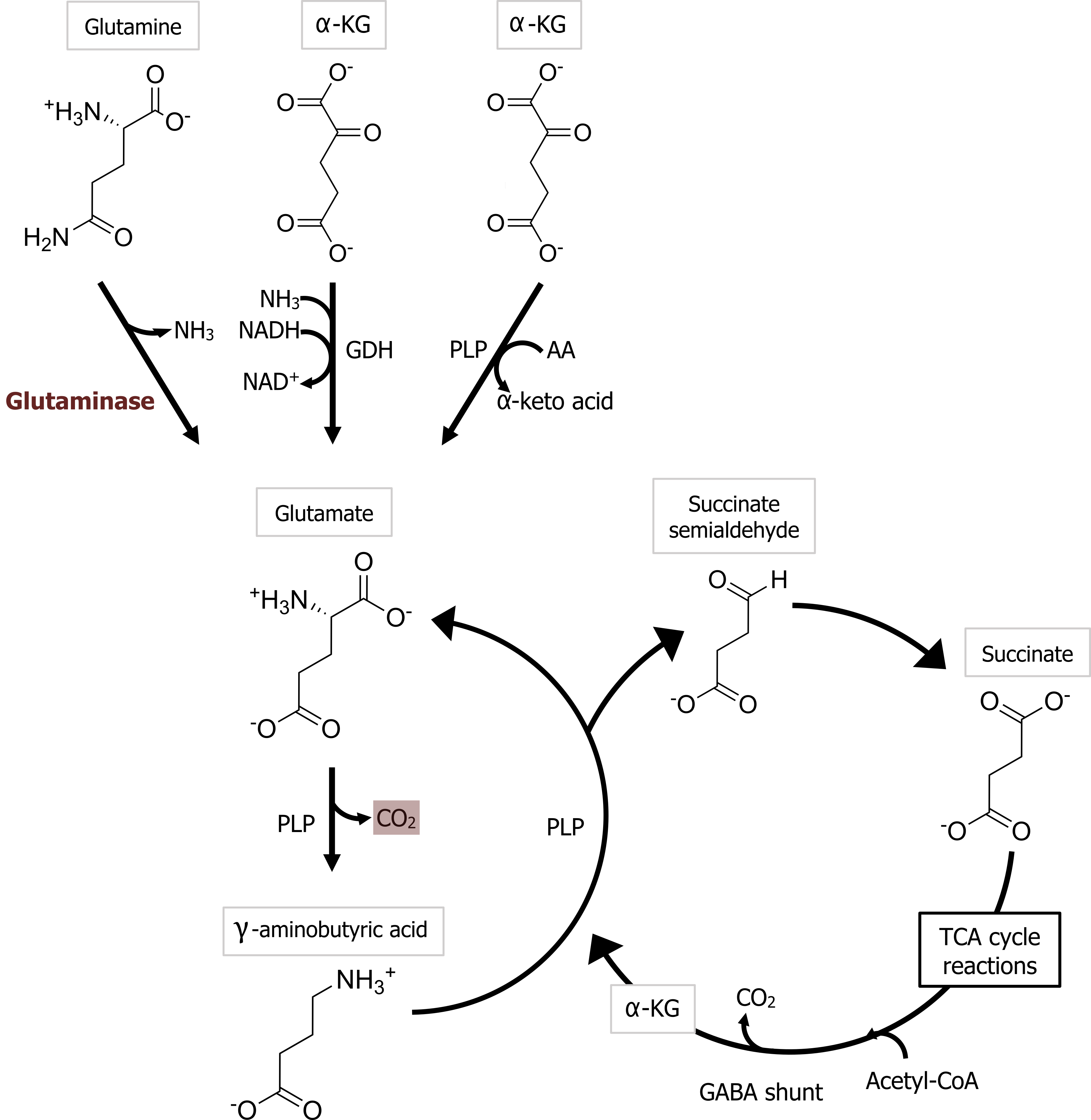
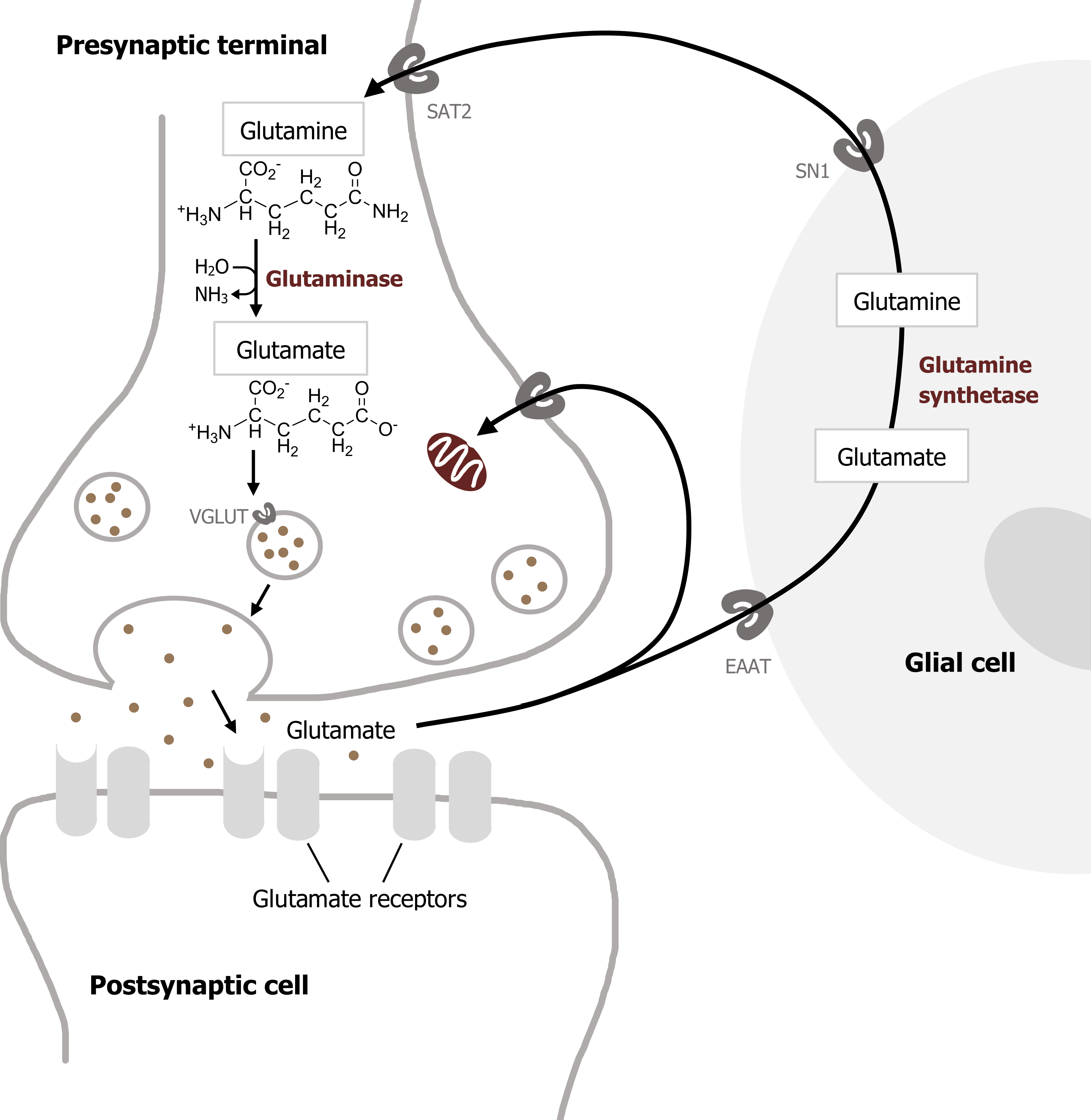
Glutamate is the most important transmitter for normal brain function. Nearly all excitatory neurons in the central nervous system (CNS) are glutamatergic. Glutamate is a nonessential amino acid that does not cross the blood-brain barrier and therefore must be synthesized in neurons from local precursors. The most prevalent precursor for glutamate synthesis is glutamine, which is taken up into presynaptic terminals by the system A transporter 2 (SAT2) and is then metabolized to glutamate by the mitochondrial enzyme glutaminase (figure 2.3).
Termination of signal
Glutamate synthesized in the presynaptic cytoplasm is packaged into synaptic vesicles by vesicular glutamate transporters (VGLUTs). Once released, glutamate is removed from the synaptic cleft by the excitatory amino acid transporters (EAATs). EAATs are a family of five different Na+-dependent glutamate cotransporters. Some EAATs are present in glial cells and others in presynaptic terminals. Glutamate transported into glial cells via EAATs is converted into glutamine by the enzyme glutamine synthetase. Glutamine is then transported out of the glial cells by a different transporter, the system N transporter 1 (SN1), and transported into nerve terminals via SAT2. This overall sequence of events is referred to as the glutamate–glutamine cycle. This cycle allows glial cells and presynaptic terminals to cooperate both to maintain an adequate supply of glutamate for synaptic transmission and to rapidly terminate postsynaptic glutamate action (figure 2.4).
Glutamate receptors
There are several types of ionotropic glutamate receptors: AMPA receptors, NMDA receptors, and kainate receptors, named after the agonists that activate them: AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate), NMDA (N-methyl-d-aspartate), and kainic acid. All of these receptors are glutamate-gated cation channels that allow the passage of Na+ and K+. Therefore AMPA, kainate, and NMDA receptor activation always produces excitatory postsynaptic responses. Most central excitatory synapses possess both AMPA and NMDA receptors. Antagonist drugs that selectively block either AMPA or NMDA receptors are often used to identify synaptic responses mediated by each receptor type. The physiological roles of kainate receptors are less well defined; in some cases, these receptors are found on presynaptic terminals and serve as a feedback mechanism to regulate glutamate release.
In addition to these ionotropic glutamate receptors, there are three classes of metabotropic glutamate receptors (mGluRs). These receptors differ in their coupling to intracellular signal transduction pathways and in their sensitivity to pharmacological agents. Activation of many of these receptors leads to inhibition of postsynaptic Ca2+ and Na+ channels. Unlike the excitatory ionotropic glutamate receptors, mGluRs cause slower postsynaptic responses that can either excite or inhibit postsynaptic cells. As a result, the physiological roles of mGluRs are quite varied.
GABA
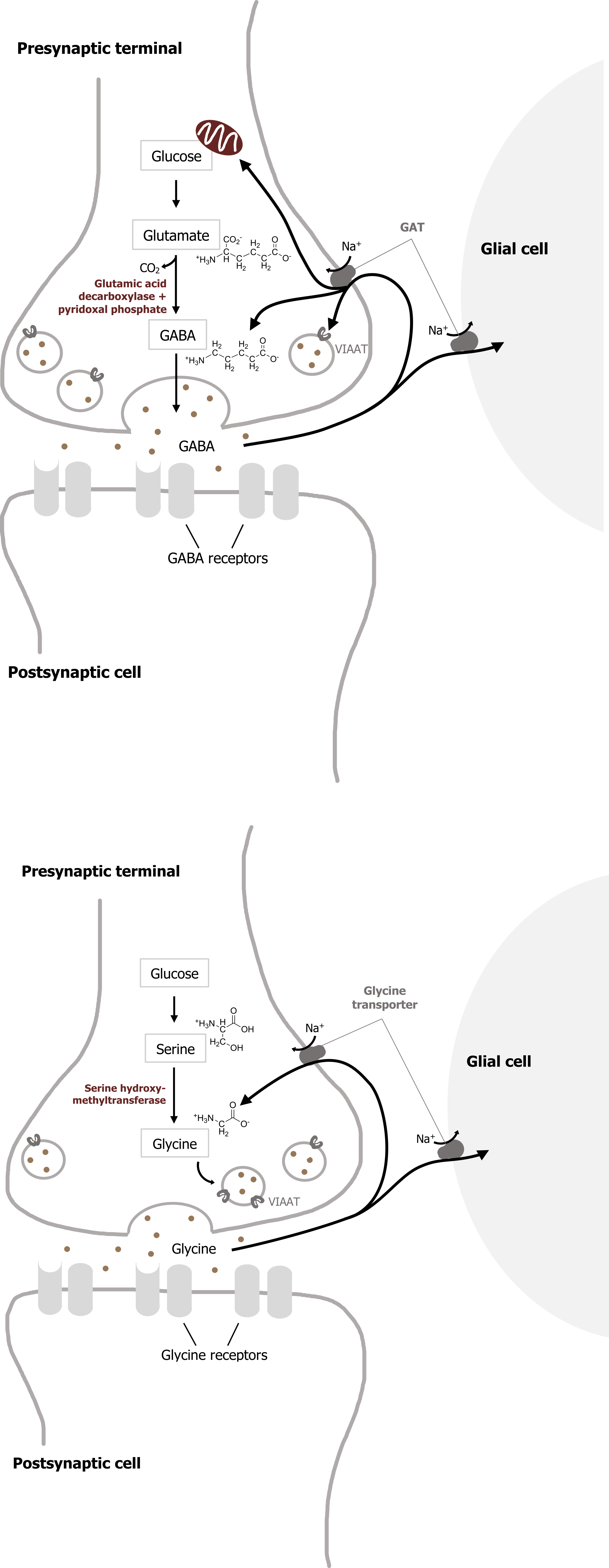
Most inhibitory synapses in the brain and spinal cord use either γ-aminobutyric acid (GABA) or glycine as neurotransmitters. The predominant precursor for GABA synthesis is glucose, which is metabolized to glutamate by the tricarboxylic acid cycle enzymes (figure 2.3). The enzyme glutamic acid decarboxylase (GAD), which is found almost exclusively in GABAergic neurons, catalyzes the conversion of glutamate to GABA. GAD requires pyridoxal phosphate for activity; a deficiency of this vitamin can lead to diminished GABA synthesis.
Termination of signal
Once GABA is synthesized, it is transported into synaptic vesicles via a vesicular inhibitory amino acid transporter (VIAAT). The mechanism of GABA removal is similar to that of glutamate. Both neurons and glia contain high-affinity Na+-dependent cotransporters for GABA, and these cotransporters are termed GATs. Most GABA is eventually converted to succinate, which is metabolized further in the tricarboxylic acid cycle that mediates cellular ATP synthesis.
Two mitochondrial enzymes are required for this degradation: GABA transaminase and succinic semialdehyde dehydrogenase.
GABA receptors
GABAergic synapses employ two types of postsynaptic receptors, called GABAA and GABAB. GABAA are ionotropic receptors, while GABAB are metabotropic receptors. The ionotropic GABAA receptors are GABA-gated anion channels, with Cl– being the main permeant ion under physiological conditions. Thus, activation of these GABA receptors causes an influx of negatively charged Cl– that inhibits postsynaptic cells. The same site binds the hypnotic zolpidem (Ambien), which is widely used to induce sleep. Barbiturates such as phenobarbital and pentobarbital are other hypnotics that also bind to the extracellular domains of the α and β subunits of some GABA receptors and potentiate GABAergic transmission; these drugs are used therapeutically for anesthesia and to control epilepsy. The injection anesthetic ketamine also binds to the extracellular domain of GABA receptors.
The transmembrane domains of GABAA receptors also serve as the targets for numerous ligands, such as inhalant anesthetics and steroids. Another drug that binds to the transmembrane domain of GABAA receptors is ethanol; at least some aspects of drunken behavior are caused by ethanol-mediated alterations in ionotropic GABA receptors.
The metabotropic GABAB receptors are also widely distributed in the brain. Like the ionotropic GABA receptors, GABAB receptors are inhibitory. Rather than relying on Cl–-selective channels, however, GABAB-mediated inhibition is often due to the activation of K+ channels and subsequent efflux of K+.
Glycine
The distribution of the neutral amino acid glycine in the CNS is more restricted than that of GABA. About half of the inhibitory synapses in the spinal cord use glycine; most other inhibitory synapses use GABA. Glycine is synthesized from serine by the mitochondrial isoform of serine hydroxymethyltransferase (figure 2.5) and is transported into synaptic vesicles via the same VIAAT that loads GABA into vesicles.
Termination of signal
Once released from the presynaptic cell, glycine is rapidly removed from the synaptic cleft by glycine transporters in the plasma membrane (figure 2.5).
Glycine receptors
Glycine receptors are pentamers consisting of a combination of four types of α subunits, along with an accessory β subunit. These receptors are potently blocked by strychnine, which may account for the toxic properties of this plant alkaloid. Glycine receptors are ligand-gated Cl– channels whose general structure closely mirrors that of the GABAA receptors.
| Neurotransmitter | Postsynaptic effect | Precursor(s) | Rate-limiting step | Removal mechanism |
|---|---|---|---|---|
| ACh | Excitatory | Chlorine + acetyl-CoA | ChAT | ACh-esterase |
| Glutamate | Excitatory | Glutamine | Glutaminase | Transporters |
| GABA | Inhibitory | Glutamine | GAD | Transporters |
| Glycine | Inhibitory | Serine | Phosphoserine | Transporters |
2.1: Acetylcholine and the primary amino acid neurotransmitters.
Biogenic amines
Biogenic amine transmitters regulate many brain functions and are also active in the peripheral nervous system. There are five well-established biogenic amine neurotransmitters, three of which can be classified as catecholamines:
1. Dopamine (catecholamine),
2. Adrenaline/noradrenaline (catecholamine),
3. Epinephrine/norepinephrine (catecholamine),
4. Histamine, and
5. Serotonin.
Dopamine
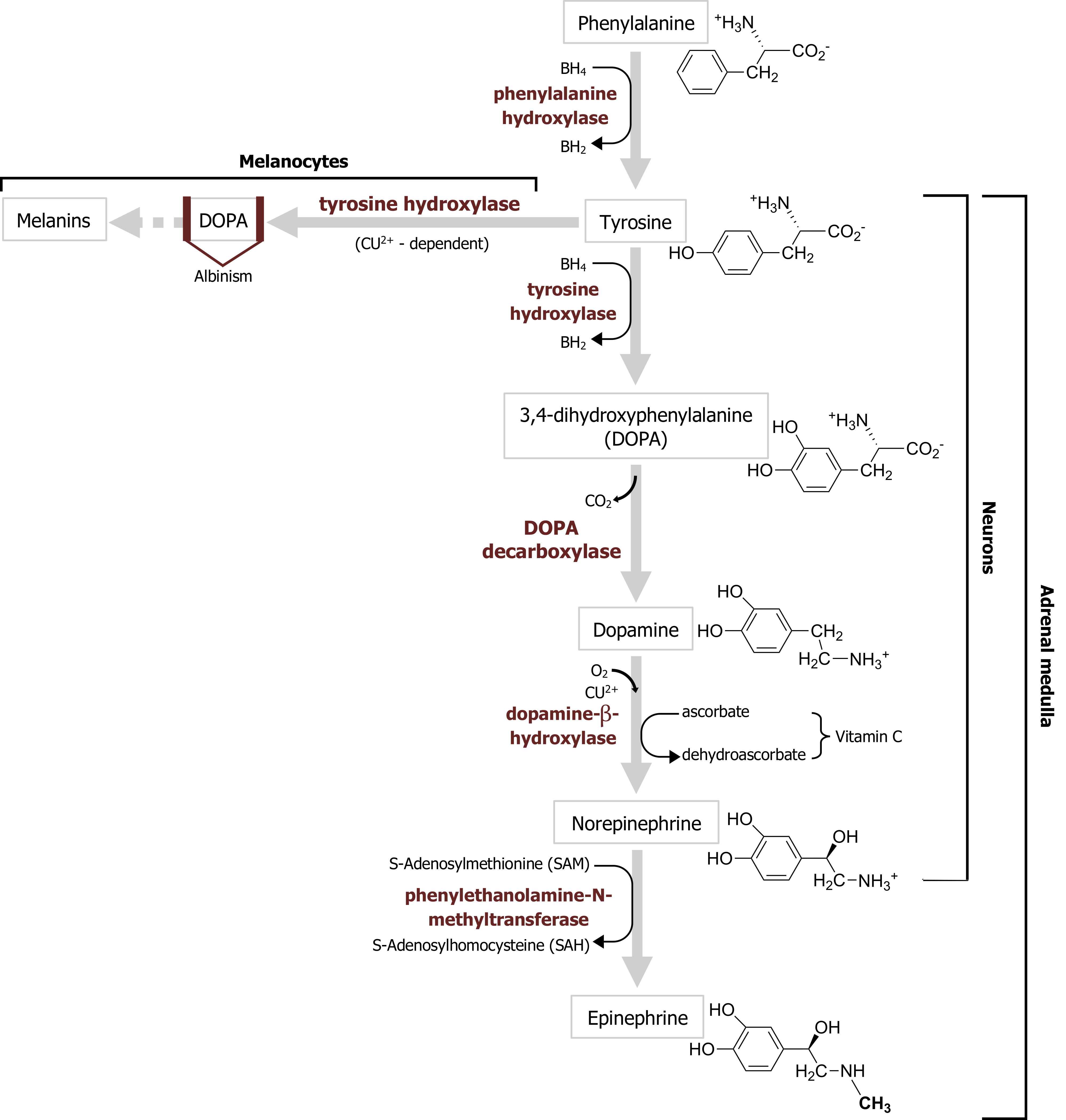
The first step in catecholamine synthesis is catalyzed by tyrosine hydroxylase in a reaction requiring oxygen as a cosubstrate and tetrahydrobiopterin as a cofactor to synthesize dihydroxyphenylalanine (DOPA) (figure 2.6).
Dopamine is produced by the action of DOPA decarboxylase on DOPA. Following its synthesis in the cytoplasm of presynaptic terminals, dopamine is loaded into synaptic vesicles via a vesicular monoamine transporter (VMAT). Dopamine action in the synaptic cleft is terminated by reuptake of dopamine into nerve terminals or surrounding glial cells by a Na+-dependent dopamine cotransporter, termed DAT.
Termination of signal
The two major enzymes involved in the catabolism of dopamine are monoamine oxidase (MAO) and catechol O-methyltransferase (COMT). Both neurons and glia contain mitochondrial MAO and cytoplasmic COMT.
Dopamine receptors
Once released, dopamine acts exclusively by activating G-protein–coupled receptors. Most dopamine receptor subtypes act by either activating or inhibiting adenylyl cyclase. Activation of these receptors generally contributes to complex behaviors.
Norepinephrine (also called noradrenaline) is used as a neurotransmitter and influences sleep and wakefulness, arousal, attention, and feeding behavior. Perhaps the most prominent noradrenergic neurons are sympathetic ganglion cells, which employ norepinephrine as the major peripheral transmitter in this division of the visceral motor system.
Norepinephrine synthesis requires dopamine β-hydroxylase, which catalyzes the production of norepinephrine from dopamine (figure 2.6). Norepinephrine is then loaded into synaptic vesicles via the same VMAT involved in vesicular dopamine transport.
Termination of signal
Norepinephrine is cleared from the synaptic cleft by the norepinephrine transporter (NET), an Na+-dependent cotransporter that is also capable of taking up dopamine. NET is a molecular target of amphetamine, which acts as a stimulant by producing a net increase in the release of norepinephrine and dopamine. Like dopamine, norepinephrine is degraded by MAO and COMT.
Epinephrine (also called adrenaline) is found in the brain at lower levels than the other catecholamines and is also present in fewer brain neurons than other catecholamines. Epinephrine-secreting neurons regulate respiration and cardiac function. The enzyme that synthesizes epinephrine, phenylethanolamine-N-methyltransferase (figure 2.6), is present only in epinephrine-secreting neurons. Otherwise, the metabolism of epinephrine is very similar to that of norepinephrine. Epinephrine is loaded into vesicles via the VMAT. No plasma membrane transporter specific for epinephrine has been identified, although the NET is capable of transporting epinephrine. As already noted, epinephrine acts on both α- and β-adrenergic receptors.
Epinephrine and norepinephrine signaling
Both norepinephrine and epinephrine act on α- and β-adrenergic receptors. Both types of receptor are G-protein–coupled; in fact, the β-adrenergic receptor was the first identified metabotropic neurotransmitter receptor. Binding of norepinephrine or epinephrine causes small changes in the structure of this receptor, which permits the G-protein to bind. This, in turn, causes larger changes in the shape of the α subunit of the G-protein, the first step in a series of reactions that allow the G-protein to regulate intracellular signaling cascades.
Two subclasses of α-adrenergic receptors have been identified. Activation of α1 – adernergic receptors usually results in a slow depolarization linked to the inhibition of K+ channels, while activation of α2 receptors produces a slow hyperpolarization due to the activation of a different type of K+ channel. There are three subtypes of β-adrenergic receptors, two of which are expressed in many types of neurons.
Histamine
Histamine is found in neurons in the hypothalamus that send sparse but widespread projections to almost all regions of the brain and spinal cord. The central histamine projections mediate arousal and attention, similar to central ACh and norepinephrine projections. Histamine also controls the reactivity of the vestibular system. Allergic reactions or tissue damage cause release of histamine from mast cells in the bloodstream. The close proximity of mast cells to blood vessels, together with the potent actions of histamine on blood vessels, raises the possibility that histamine may influence brain blood flow.
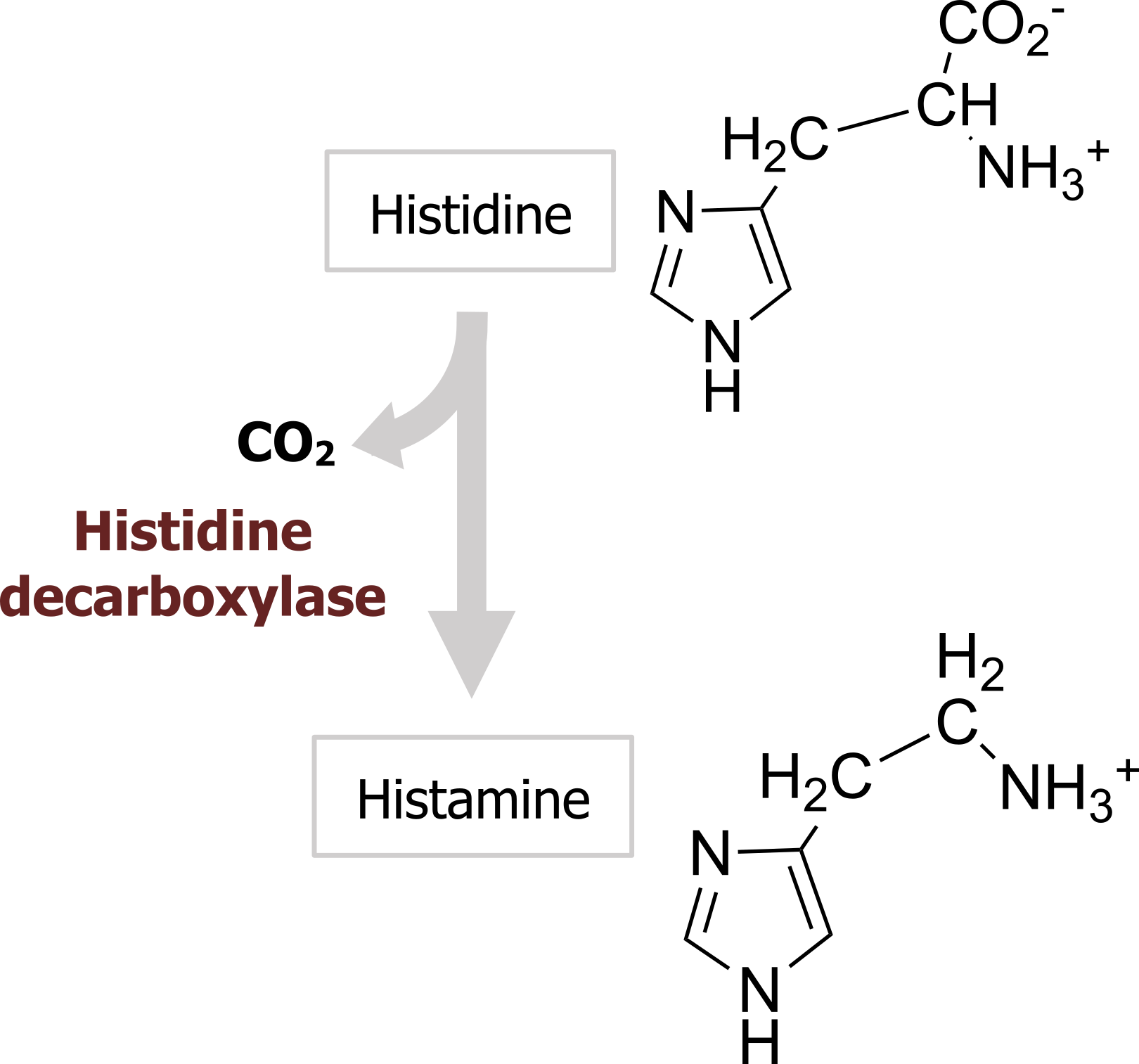
Histamine is produced from the amino acid histidine by a histidine decarboxylase (figure 2.7) and is transported into vesicles via the same VMAT as the catecholamines. No plasma membrane histamine transporter has been identified yet.
Termination of signal
Histamine is degraded by the combined actions of histamine methyltransferase and MAO.
Histamine receptors
The four known types of histamine receptors are all metabotropic receptors. Of the four, only two of the receptors (H1 and H2) are well characterized. Because of the role of histamine receptors in mediating allergic responses, many histamine receptor antagonists have been developed as antihistamine agents. Antihistamines that cross the blood-brain barrier, such as diphenhydramine (Benadryl), act as sedatives by interfering with the roles of histamine in CNS arousal. Antagonists of the H1 receptor also are used to prevent motion sickness, perhaps because of the role of histamine in controlling vestibular function (figure 2.8).

H2 receptors control the secretion of gastric acid in the digestive system, allowing H2 receptor antagonists to be used in treating a variety of upper gastrointestinal disorders (e.g., peptic ulcers).
Serotonin
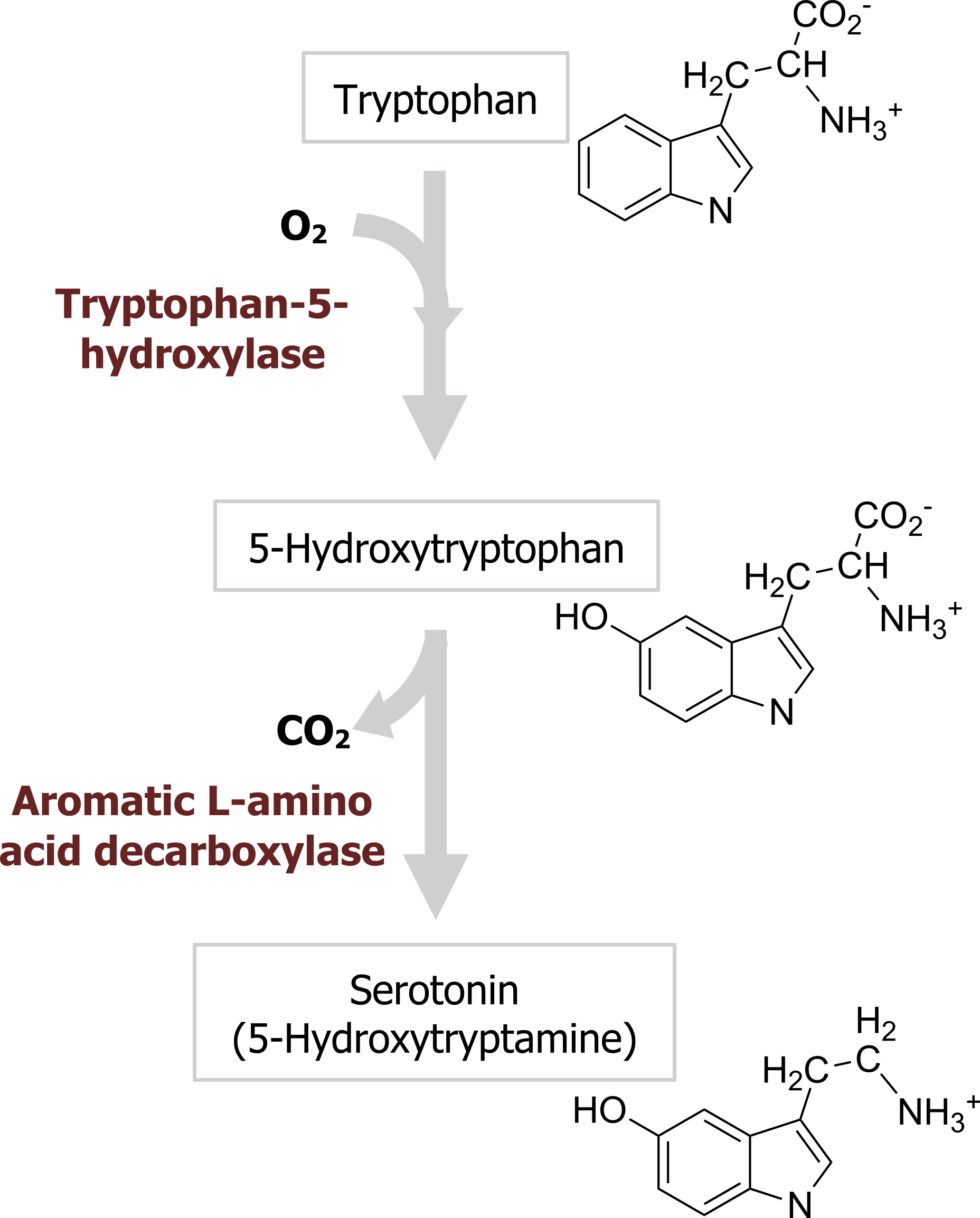
Serotonin, or 5-hydroxytryptamine (5-HT), was initially thought to increase vascular tone by virtue of its presence in blood serum (hence the name serotonin). 5-HT is synthesized from the amino acid tryptophan, which is an essential dietary requirement. Tryptophan is taken up into neurons by a plasma membrane transporter and hydroxylated in a reaction catalyzed by the enzyme tryptophan-5-hydroxylase (figure 2.9), the rate-limiting step for 5-HT synthesis. Loading of 5-HT into synaptic vesicles is done by the VMAT that is also responsible for loading other monoamines into synaptic vesicles. The synaptic effects of serotonin are terminated by transport back into nerve terminals via a specific serotonin transporter (SERT) that is present in the presynaptic plasma membrane and is encoded by the 5HTT gene. Many antidepressant drugs are selective serotonin reuptake inhibitors (SSRIs) that inhibit transport of 5-HT by SERT. Perhaps the best-known example of an SSRI is the antidepressant drug Prozac.
Termination of signal
The primary catabolic pathway for 5-HT is mediated by MAO.
Serotonin receptors
Most 5-HT receptors are metabotropic, with a monomeric structure typical of G-protein–coupled receptors. Metabotropic 5-HT receptors have been implicated in a wide range of behaviors, including circadian rhythms, motor behaviors, emotional states, and mental arousal. Impairments in the function of these receptors have been implicated in numerous psychiatric disorders, such as depression, anxiety disorders, and schizophrenia, and drugs acting on serotonin receptors are effective treatments for several of these conditions.
| Neurotransmitter | Postsynaptic effect | Precursor(s) | Rate-limiting step | Removal mechanism |
|---|---|---|---|---|
| ACh | Excitatory | Choline + acetyl-CoA | ChAT | ACh-esterase |
| Glutamate | Excitatory | Glutamine | Glutaminase | Transporters |
| GABA | Inhibitory | Glutamine | GAD | Transporters |
| Glycine | Inhibitory | Serine | Phosphoserine | Transporters |
| Catecholamines (dopamine, epi, norepi) | Excitatory | Tyrosine | Tyrosine hydroxylase | MAO; COMT |
| Histamine | Excitatory | Histidine | Histidine decarboxylase | Transporters |
| Serotonin (5-HT) | Excitatory | Tryptophan | Tryptophan hydroxylase | MAO; COMT |
Table 2.2: Summary of neurotransmitter synthesis and degradation.
References and resources
Text
“Biochemistry of Nerve Transmission.” The Medical Biochemistry Page. http://themedicalbiochemistrypage.or…-transmission/.
“Neurotransmitters and Receptors: Different Classes of Neurotransmitters, and Different Types of Receptors They Bind To.” Khan Academy. https://www.khanacademy.org/science/…heir-receptors.
Purves, D., G. J. Augustine, Dd. Fitzpatrick, L. C. Katz, A.-S. LaMantia, J. O. McNamara, and S. M. Williams, eds. Neuroscience, 2nd ed. Sunderland, MA: Sinauer Associates, 2001, Chapter 6: Neurotransmitters.
Figures
Figure 2.1: Synthesis and degradation of acetylcholine. Grey, Kindred. “Synthesis and Degradation of Acetylcholine.” 2021, https://archive.org/details/2.1_20210817. CC BY 4.0. Chemical structure by Henry Jakubowski.
Figure 2.2: ACh release and degradation. Grey, Kindred. “ACh Release and Degradation.” 2021, https://archive.org/details/2.2_20210817. CC BY 4.0. Added curved left arrow by Star and Anchor Design from the Noun Project.
Figure 2.3: Glutamate and GABA synthesis. Lieberman, M., and A. Peet, eds. Marks’ Basic Medical Biochemistry: A Clinical Approach, 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2018, 747, Figure 35.7: The gama glutamyl cycle. Adapted under fair use. Chemical structure by Henry Jakubowski.
Figure 2.4: Glutamate release and reuptake. Purves, D., G. J. Augustine, D. Fitzpatrick, W. C. Hall, A.-S. LaMantia, R. D. Mooney, M. L. Platt, and L. E. White. Neuroscience, 6th ed. New York: Oxford University Press, 2017, 122, Figure 6.5: Glutamate synthesis and cycling between neurons and glia. Adapted under fair use. Added beans by Vectors Market from the Noun Project. Chemical structure by Henry Jakubowski.
Figure 2.5: GABA and glycine release. Purves, D., G. J. Augustine, D. Fitzpatrick, W. C. Hall, A.-S. LaMantia, R. D. Mooney, M. L. Platt, and L. E. White. Neuroscience, 6th ed. New York: Oxford University Press, 2017, 127, Figure 6.10: Synthesis, release and reuptake of the inhibitor neurotransmitters GABA and glycine. Adapted under fair use. Added beans by Vectors Market from the Noun Project. Chemical structure by Henry Jakubowski.
Figure 2.6: Synthesis of dopamine, norepinephrine, and epinephrine. Grey, Kindred. “Synthesis of Dopamine, Norepinephrine and Epinephrine. 2021, https://archive.org/details/2.6_20210819. CC BY 4.0. Chemical structure by Henry Jakubowski.
Figure 2.7: Histamine synthesis. Purves, D., G. J. Augustine, D. Fitzpatrick, W. C. Hall, A.-S. LaMantia, R. D. Mooney, M. L. Platt, and L. E. White. Neuroscience, 6th ed. New York: Oxford University Press, 2017, 134, Figure 6.18: Synthesis of histamine and serotonin. Adapted under fair use. Chemical structure by Henry Jakubowski.
Figure 2.8: Histamine release and reuptake. Grey, Kindred. “Histamine Release and Reuptake.” 2021, https://archive.org/details/2.8_20210819. CC BY 4.0.
Figure 2.9: Serotonin synthesis. Purves, D., G. J. Augustine, D. Fitzpatrick, W. C. Hall, A.-S. LaMantia, R. D. Mooney, M. L. Platt, and L. E. White. Neuroscience, 6th ed. New York: Oxford University Press, 2017, 134, Figure 6.18: Synthesis of histamine and serotonin. Adapted under fair use. Chemical structure by Henry Jakubowski.
