9 Foundations of Disease Management
9.1 Fundamentals of Antimicrobial Chemotherapy
Several factors are important in choosing the most appropriate antimicrobial drug therapy, including bacteriostatic versus bactericidal mechanisms, spectrum of activity, dosage and route of administration, the potential for adverse side effects, and the potential interactions between drugs. The following discussion will focus primarily on antibacterial drugs, but the concepts translate to other antimicrobial classes.
Bacteriostatic Versus Bactericidal
Antibacterial drugs can be classified as either bacteriostatic or bactericidal in their interactions with target bacteria. It is important to note that both types, such as tetracyclines and macrolides (bacteriostatic), and beta-lactams and fluoroquinolones (bactericidal) kill bacteria. However, bacteriostatic drugs require a higher concentration than bactericidal agents to achieve specific bacterial reduction. A literature review utilizing data from randomized control trials demonstrated there is no superiority of bactericidal agents to bacteriostatic agents. Alternative factors such as drug pharmacokinetics, therapeutic dosing, and tissue penetration, serve as more significant efficacy determinants.[1]
Spectrum of Activity
The spectrum of activity of an antibacterial drug relates to diversity of targeted bacteria. A narrow-spectrum antimicrobial targets only specific subsets of bacterial pathogens. For example, some narrow-spectrum drugs only target gram-positive bacteria, whereas others target only gram-negative bacteria. If the pathogen causing an infection has been identified, it is best to use a narrow-spectrum antimicrobial and minimize collateral damage to the normal microbiota. A broad-spectrum antimicrobial targets a wide variety of bacterial pathogens, encompassing both gram-positive and gram-negative species. They are often employed as empiric therapy to cover a wide range of potential pathogens while waiting for the laboratory identification of the infecting pathogen. Broad-spectrum antimicrobials are also used for polymicrobial infections (mixed infection with multiple bacterial species), or as prophylactic measures prior to surgery or invasive procedures. Finally, broad-spectrum antimicrobials may be selected to treat an infection when a narrow-spectrum drug fails because of development of drug resistance by the target pathogen.
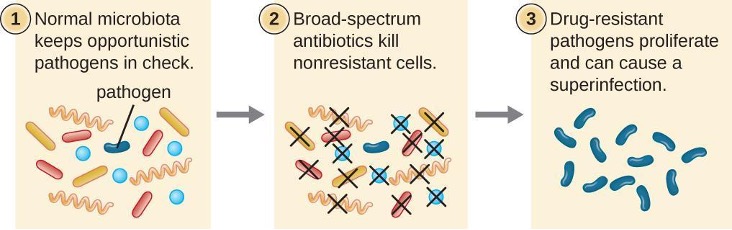
The risk associated with using broad-spectrum antimicrobials is that they will also target a broad spectrum of the normal microbiota, increasing the risk of a superinfection, a secondary infection in a patient having a preexisting infection. Superinfections develop when the antibacterial intended for the preexisting infection kills the protective microbiota, allowing another pathogen resistant to the antibacterial to proliferate and cause a secondary infection (figure 9.1). Common superinfections that develop from appropriate and inappropriate antimicrobial utilization include yeast infections (candidiasis) and pseudomembranous colitis caused by Clostridium difficile.
Dosage and Route of Administration
The dosage of a medication, defined as the amount administered within a specific time interval, requires careful determination to achieve optimal therapeutic drug levels at the infection site while minimizing potential toxicity (side effects) for the patient. Each drug class is associated with a variety of potential side effects, and some of these are described for specific drugs later in this chapter. Despite best efforts to optimize dosing, allergic reactions and other potentially serious side effects are not uncommon. Therefore, the goal is to select the optimum dosage that will minimize the risk of side effects while still achieving clinical cure, and there are important factors to consider when selecting the best dose and dosage interval.
For example, in pediatric patients, the dose is typically based upon the patient’s mass. In adult patients, antimicrobial dosing varies depending on the specific agent used, and it may not always follow standard dosing protocols. Factors such as total, ideal, or adjusted body weight may be considered in determining the dose for some antimicrobials, while others may involve flat dosing. With the great variability in adult body mass, some experts have argued that mass should be considered for all patients when determining appropriate dosage.[2] Additionally, renal and hepatic function play a crucial role in determining the appropriate dose and frequency of the dosing interval. In general, patients with a history of liver or kidney dysfunction may experience reduced drug metabolism or clearance from the body, resulting in increased drug levels that may lead to toxicity and increase their susceptibility to side effects. Furthermore, certain antimicrobial agents, such as vancomycin and aminoglycosides, may require serum concentration monitoring to accurately guide subsequent dosing regimens.
There are also some factors specific to the drugs themselves that influence appropriate dose and time interval between doses. For example, the half-life, or rate at which 50% of a drug is eliminated from the plasma, can vary significantly between drugs. Some drugs have a short half-life of only 1 hour and must be given multiple times a day, whereas other drugs have half-lives exceeding 12 hours and can be given as a single dose every 24 hours. While a longer half-life may offer the advantage of convenient dosing intervals, it can be a concern for drugs with serious side effects, as elevated drug levels may persist for an extended period of time. Additionally, some drugs are dose dependent, meaning they are more effective when administered in large doses to provide high levels for a short time at the site of infection. Others are time dependent, meaning they are more effective when lower drug levels are maintained over the minimum inhibitory concentration for a longer period of time.
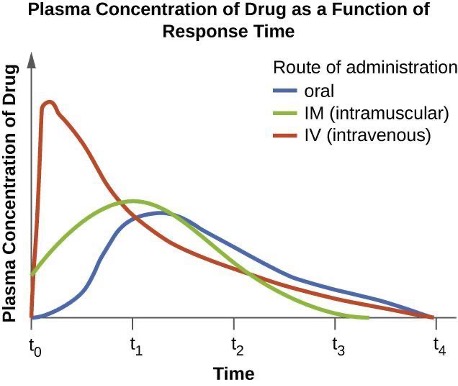
The route of administration, the method used to introduce a drug into the body, is also an important consideration for drug therapy. Oral administration is generally preferred for its convenience, allowing patients to self-administer at home. However, some drugs are poorly absorbed from the gastrointestinal (GI) tract into the bloodstream. These drugs are often useful for treating diseases of the intestinal tract, such as tapeworms treated with praziquantel, or for ulcerative colitis, as with sulfasalazine. Some drugs that are not absorbed easily, such as bacitracin, polymyxin, and several antifungals, are available as topical preparations for treatment of superficial skin infections. In cases where oral administration is initially challenging due to illness or other factors, parenteral routes (intravenous or intramuscular) are preferred in healthcare settings. Intravenous administration can result in higher plasma levels compared to other routes of administration and oftentimes is a consideration for therapy selection. It is important to recognize that certain oral formulations exhibit identical bioavailability to their intravenous counterparts (e.g. linezolid, metronidazole, levofloxacin) and this should be taken into account when selecting the formulation (figure 9.2).
Drug Interactions
For the optimal treatment of certain infections, a combination of two antibacterial drugs may be administered together to achieve a synergistic interaction that surpasses the efficacy of either drug alone. A classic example of synergistic combinations is trimethoprim and sulfamethoxazole (Bactrim®). Individually, these two drugs provide only bacteriostatic inhibition of bacterial growth, but combined, the drugs are bactericidal.
Whereas synergistic drug interactions provide a benefit to the patient, antagonistic interactions produce harmful effects. Antagonism can occur between two antimicrobials or between antimicrobials and non-antimicrobials being used to treat other conditions. The effects vary depending on the drugs involved, but antagonistic interactions may cause loss of drug activity, decreased therapeutic levels due to increased metabolism and elimination, or increased potential for toxicity due to decreased metabolism and elimination. As an example, some antibacterials are absorbed most effectively from the acidic environment of the stomach. If a patient takes antacids, however, this increases the pH of the stomach and negatively impacts the absorption of these antimicrobials, decreasing their effectiveness in treating an infection.[3]
9.2 Mechanisms of Antibacterial Drugs
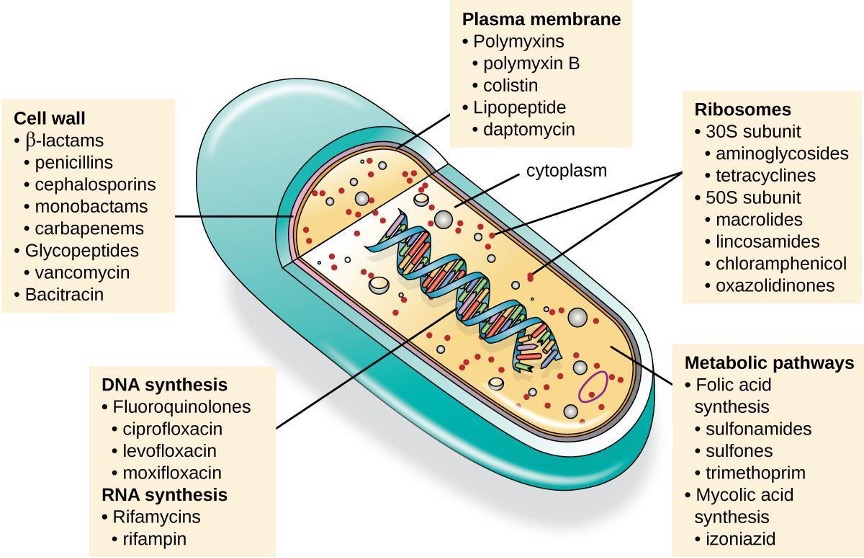
An essential attribute for an antimicrobial drug is selective toxicity, meaning that it selectively kills or inhibits the growth of microbial targets while causing minimal or no harm to the host. Most antimicrobial drugs currently in clinical use are antibacterial because the prokaryotic cell provides a greater variety of unique targets for selective toxicity, in comparison to fungi, parasites, and viruses. Each class of antibacterial drugs has a unique mode of action (the way in which a drug affects microbes at the cellular level), and these are summarized in figure 9.3 and table 9.1.
| Mode of Action | Target | Drug Class |
|---|---|---|
| Inhibit cell wall biosynthesis | Penicillin-binding proteins | β-lactams: penicillins, cephalosporins, monobactams, carbapenems |
| Peptidoglycan subunits | Glycopeptides | |
| Peptidoglycan subunit transport | Bacitracin | |
| Inhibit biosynthesis of proteins | 30S ribosomal subunit | Aminoglycosides, tetracyclines |
| 50S ribosomal subunit | Macrolides, lincosamides, chloramphenicol, oxazolidinones | |
| Disrupt membranes | Lipopolysaccharide, inner and outer membranes | Polymyxin B, colistin, daptomycin |
| Inhibit nucleic acid synthesis | RNA | Rifamycin |
| DNA | Fluoroquinolones | |
| Antimetabolites | Folic acid synthesis enzyme | Sulfonamides, trimethoprim |
| Mycolic acid synthesis enzyme | Isoniazid |
Table 9.1: Common antibacterial drugs by mode of action
Inhibitors of Cell Wall Biosynthesis
Several classes of antibacterials, such as beta-lactam antibiotics (e.g., penicillins, cephalosporins) and glycopeptide antibiotics (e.g., vancomycin), target specific steps in the biosynthesis of peptidoglycan, a crucial component of bacterial cell walls. By inhibiting peptidoglycan formation, these antibacterials render bacterial cells more susceptible to osmotic lysis (table 9.2). Because human cells do not make peptidoglycan, this mode of action is an excellent example of selective toxicity.
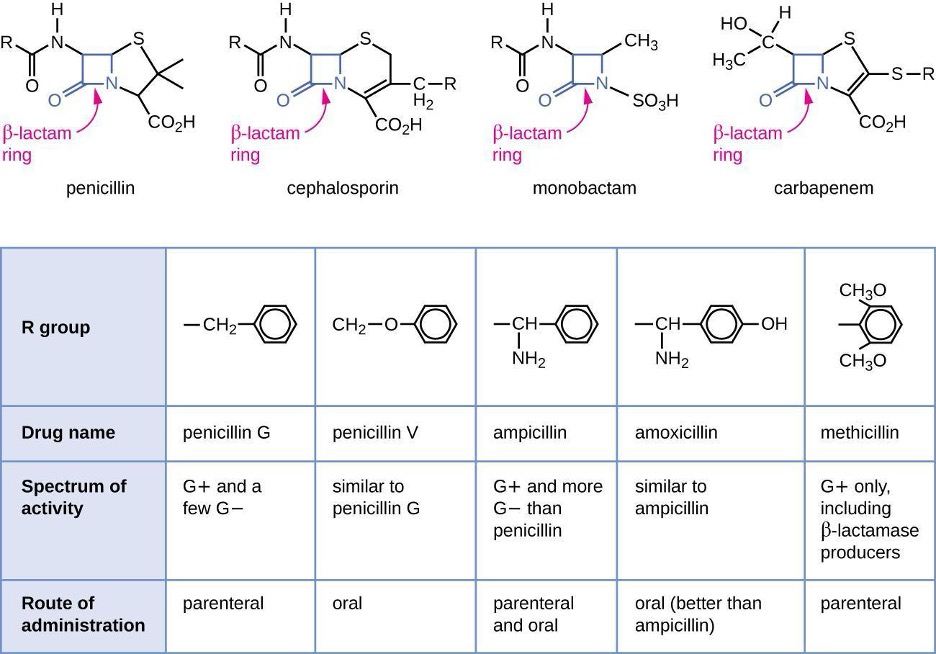
Penicillin, the first antibiotic discovered, is one of several antibacterials within a class called β-lactams. This group of compounds includes the penicillins, cephalosporins, monobactams, and carbapenems, and is characterized by the presence of a β-lactam ring found within the central structure of the drug molecule (figure 9.4). The β-lactam antibacterials block the crosslinking of peptide chains during the biosynthesis of new peptidoglycan in the bacterial cell wall. They can block this process because the β-lactam structure is similar to the structure of the peptidoglycan subunit component that is recognized by the crosslinking transpeptidase enzyme, also known as a penicillin-binding protein (PBP). Although the β-lactam ring must remain unchanged for these drugs to retain their antibacterial activity, strategic chemical changes to the R groups have allowed for development of a wide variety of semisynthetic β-lactam drugs with increased potency, expanded spectrum of activity, and longer half-lives for better dosing, among other characteristics.
Penicillin G and penicillin V are natural antibiotics from fungi and are primarily active against gram-positive bacterial pathogens, and a few gram-negative bacterial pathogens. Penicillinase-resistant penicillins, including dicloxacillin and oxacillin, are a subgroup of antibiotics designed to withstand the enzymatic degradation by beta-lactamases produced by certain bacteria. These antibiotics maintain efficacy against penicillinase-producing strains of bacteria, making them valuable in the treatment of staphylococcal infections, including skin and soft tissue infections.
Similar to the penicillins, cephalosporins contain a β-lactam ring (figure 9.4) and block the transpeptidase activity of penicillin-binding proteins. However, the β-lactam ring of cephalosporins is fused to a six-member ring, rather than the five-member ring found in penicillins. This chemical difference provides cephalosporins with an increased resistance to enzymatic inactivation by β-lactamases. The family of semisynthetic cephalosporins is much larger than the penicillins, and these drugs have been classified into generations based primarily on their spectrum of activity, increasing in spectrum from the narrow-spectrum, first-generation cephalosporins to the broad-spectrum, fourth generation cephalosporins. Ceftaroline and ceftolozane are new fifth-generation cephalosporins. Ceftaroline has activity against methicillin-resistant Staphylococcus aureus (MRSA) while ceftolozane provides extended Pseudomonas aeruginosa coverage.
The carbapenems and monobactams also have a β-lactam ring as part of their core structure, and they inhibit the transpeptidase activity of penicillin-binding proteins. The only monobactam used clinically is aztreonam. It has activity only against gram-negative bacteria, including Pseudomonas aeruginosa. In contrast, the carbapenem family includes a variety of semisynthetic drugs (imipenem, meropenem, and doripenem) that provide very broad-spectrum activity against gram-positive and gram-negative bacterial pathogens.
The drug vancomycin, a member of a class of compounds called glycopeptides, was discovered in the 1950s as a natural antibiotic from the actinomycete Amycolatopsis orientalis. Similar to the β-lactams, vancomycin inhibits cell wall biosynthesis and is bactericidal. However, in contrast to the β-lactams, the structure of vancomycin is not similar to that of cell-wall peptidoglycan subunits and does not directly inactivate penicillin-binding proteins. Rather, vancomycin is a very large, complex molecule that binds to the end of the peptide chain of cell wall precursors. This creates a structural blockage that prevents the cell wall subunits from being incorporated into the growing N-acetylglucosamine and N-acetylmuramic acid (NAM-NAG) backbone of the peptidoglycan structure (transglycosylation). Vancomycin also structurally blocks transpeptidation. Vancomycin is bactericidal against gram-positive bacterial pathogens, but it is not active against gram-negative bacteria because of its inability to penetrate the protective outer membrane.
The drug bacitracin consists of a group of structurally similar peptide antibiotics originally isolated from Bacillus subtilis. Bacitracin blocks the activity of a specific cell-membrane molecule that is responsible for the movement of peptidoglycan precursors from the cytoplasm to the exterior of the cell, ultimately preventing their incorporation into the cell wall. Bacitracin is effective against a wide range of bacteria, including gram-positive organisms found on the skin, such as Staphylococcus and Streptococcus. Although it may be administered orally or intramuscularly in some circumstances, bacitracin has been shown to be nephrotoxic (damaging to the kidneys). Therefore, it is more commonly combined with neomycin and polymyxin in topical ointments such as Neosporin®.
| Mechanism of Action | Drug Class | Specific Drugs | Spectrum of Activity |
|---|---|---|---|
| Interact directly with PBPs and inhibit transpeptidase activity | Penicillins | Penicillin G, penicillin V | Streptococci, basic anaerobes (e.g.,non-difficile Clostridioides spp), syphilis (Treponema pallidum) |
| Ampicillin, amoxicillin | Enterococci, Streptococci, basic anaerobes, Listeria monocytogenes, | ||
| Cephalosporins | First generation: cefazolin cephalexin cefadroxil |
Coverage against most gram-positive cocci, methicillin susceptible Staphylococcus aureus (MSSA) as well as some gram-negative (E. Coli, P. mirabilis, K. pneumononiae) | |
| Second generation: cefuroxime cefoxitin cefotetan cefaclor cefprozil |
Coverage against streptococci and MSSA with increased gram-negative coverage Haemophilus influenzae, Neisseria, Proteus, E. coli, Klebsiella |
||
| Third generation: ceftriaxone ceftazidime cefdinir cefpodoxime cefixime cefotaxime ceftibuten Fourth generation: |
Streptococci (better than 1st or 2nd gen), MSSA (not as good as 1st or 2nd gen), increased gram-negative coverage (ceftazidime has Pseudomonas activity)
Streptococci (better than 1st or 2nd gen), MSSA, increased Gram-negative coverage (better than 3rd gen) and has Pseudomonas activity |
||
| Fifth generation cephalosporin: ceftaroline ceftolozane |
Activity against gram-positive and gram-negative bacteria
Ceftaroline provides MRSA coverage Ceftolozane provides extended Pseudomonas aeruginosa coverage |
||
| Monobactams | Aztreonam | Gram-negative coverage including Pseudomonas aeruginosa | |
| Carbapenems | Imipenem, meropenem, doripenem, ertapenem |
MSSA, streptococci, anaerobes (including B. fragilis) ertapenem lacks coverage for Acinetobacter, Pseudomonas, Enterococcus |
|
| Large molecules that bind to the peptide chain of peptidoglycan subunits, blocking transglycosylation and transpeptidation | Glycopeptides | Vancomycin | Gram-positive bacteria streptococci, MRSA, enterococci |
| Block transport of peptidoglycan subunits across cytoplasmic membrane | Bacitracin | Bacitracin | Gram-positive and gram-negative bacteria |
Table 9.2: Drugs that inhibit bacterial cell wall synthesis
Inhibitors of Protein Biosynthesis
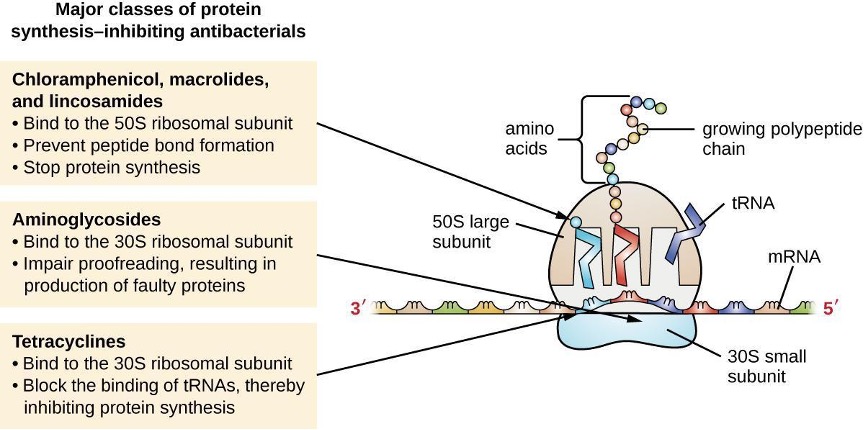
The cytoplasmic ribosomes found in animal cells (80S) are structurally distinct from those found in bacterial cells (70S), making protein biosynthesis a good selective target for antibacterial drugs. Several types of protein biosynthesis inhibitors are discussed in this section and are summarized in figure 9.5.
Protein Synthesis Inhibitors That Bind the 30S Subunit
Aminoglycosides are large, highly polar antibacterial drugs that bind to the 30S subunit of bacterial ribosomes, impairing the proofreading ability of the ribosomal complex. This impairment causes mismatches between codons and anticodons, resulting in the production of proteins with incorrect amino acids and shortened proteins that insert into the cytoplasmic membrane. Disruption of the cytoplasmic membrane by the faulty proteins kills the bacterial cells. The aminoglycosides, which include drugs such as streptomycin, gentamicin, neomycin, amikacin and tobramycin, are potent broad-spectrum antibacterials. However, aminoglycosides have been shown to be nephrotoxic (damaging to kidney), neurotoxic (damaging to the nervous system), and ototoxic (damaging to the ear).
Another class of antibacterial compounds that bind to the 30S subunit are the tetracyclines. In contrast to aminoglycosides, these drugs are bacteriostatic and inhibit protein synthesis by blocking the association of tRNAs with the ribosome during translation. Naturally occurring tetracyclines produced by various strains of Streptomyces were first discovered in the 1940s, and several semisynthetic tetracyclines, including doxycycline, and minocycline have also been produced. Although the tetracyclines are broad spectrum in their coverage of bacterial pathogens, side effects that can limit their use include phototoxicity, permanent discoloration of developing teeth, and liver toxicity with high doses or in patients with kidney impairment.
Protein Synthesis Inhibitors That Bind the 50S Subunit
There are several classes of antibacterial drugs that work through binding to the 50S subunit of bacterial ribosomes. The macrolide antibacterial drugs have a large, complex ring structure and are part of a larger class of naturally produced secondary metabolites called polyketides, complex compounds produced in a stepwise fashion through the repeated addition of two-carbon units by a mechanism similar to that used for fatty acid synthesis. Macrolides are broad-spectrum, bacteriostatic drugs that block elongation of proteins by inhibiting peptide bond formation between specific combinations of amino acids. The first macrolide was erythromycin. It was isolated in 1952 from Streptomyces erythreus and prevents translocation. Semisynthetic macrolides include azithromycin and clarithromycin. Compared with erythromycin, azithromycin has a broader spectrum of activity, fewer side effects, and a significantly longer half-life (1.5 hours for erythromycin versus 68 hours for azithromycin) that allows for once-daily dosing and a shorter course of therapy (i.e., Z-Pak® formulation) for most infections. The lincosamides include the naturally produced lincomycin and semisynthetic clindamycin. Although structurally distinct from macrolides, lincosamides are similar in their mode of action to the macrolides through binding to the 50S ribosomal subunit and preventing peptide bond formation. Lincosamides are particularly active against streptococcal and staphylococcal infections.
The drug chloramphenicol represents yet another structurally distinct class of antibacterials that also bind to the 50S ribosome, inhibiting peptide bond formation. Chloramphenicol, produced by Streptomyces venezuelae, was discovered in 1947; in 1949, it became the first broad-spectrum antibiotic that was approved by the FDA. Although it is a natural antibiotic, it is also easily synthesized and was the first antibacterial drug synthetically mass produced. As a result of its mass production, broad-spectrum coverage, and ability to penetrate tissues efficiently, chloramphenicol was historically used to treat a wide range of infections, from meningitis to typhoid fever to conjunctivitis. Unfortunately, serious side effects, such as the lethal gray baby syndrome, and suppression of bone marrow production, have limited its clinical role. Chloramphenicol also causes anemia in two different ways. One mechanism involves the targeting of mitochondrial ribosomes within hematopoietic stem cells, causing a reversible, dose-dependent suppression of blood cell production. Once chloramphenicol dosing is discontinued, blood cell production returns to normal. This mechanism highlights the similarity between 70S ribosomes of bacteria and the 70S ribosomes within our mitochondria. The second mechanism of anemia is idiosyncratic (i.e., the mechanism is not understood), and involves an irreversible lethal loss of blood cell production known as aplastic anemia. This mechanism of aplastic anemia is not dose dependent and can develop after therapy has stopped. Because of toxicity concerns, chloramphenicol usage in humans is now rare in the United States and is limited to severe infections unable to be treated by less toxic antibiotics. Because its side effects are much less severe in animals, it is used in veterinary medicine.
The oxazolidinones, including linezolid and tedizolid, are a newer broad-spectrum class of synthetic protein synthesis inhibitors that bind to the 50S ribosomal subunit of gram-positive bacteria. However, their mechanism of action seems somewhat different from that of the other 50S subunit-binding protein synthesis inhibitors already discussed. Instead, they seem to interfere with formation of the initiation complex (association of the 50S subunit, 30S subunit, and other factors) for translation, and they prevent translocation of the growing protein from the ribosomal A site to the P site. Table 9.3 summarizes the protein synthesis inhibitors.
| Molecular Target | Mechanism of Action | Drug Class | Specific Drugs | Spectrum of Activity |
|---|---|---|---|---|
| 30S subunit | Causes mismatches between codons and anticodons, leading to faulty proteins that insert into and disrupt cytoplasmic membrane | Aminoglycosides | Streptomycin, gentamicin, neomycin, amikacin, tobramycin |
Extensive gram-negative coverage; some gram-positive coverage (not adequate as monotherapy) |
| Blocks association of tRNAs with ribosome | Tetracyclines | Tetracycline, doxycycline, minocycline |
Gram-positive and gram-negative with activity against many atypical pathogens (e.g. Rickettsia spp, Chlamydia spp.) | |
| 50S subunit | Blocks peptide bond formation between amino acids | Macrolides | Erythromycin, azithromycin, clarithromycin | Many gram-negative, atypical, and mycobacterial organisms as well as gram-positive organisms |
| Lincosamides | Lincomycin, clindamycin | staphylococci, viridans group streptococci, Streptococcus pyogenes, and Streptococcus pneumoniae, anaerobes (e.g. Clostridium perfringens) | ||
| Not applicable | Chloramphenicol | gram-positive and gram-negative | ||
| Interferes with the formation of the initiation complex between 50S and 30S subunits and other factors. | Oxazolidinones | Linezolid, tedizolid | Gram-positive bacteria including streptococci, enterococci (including vancomycin-resistant enterococci [VRE]), coagulase-negative staphylococci, MSSA, MRSA, Bacillus species, Corynebacterium species, and Listeria monocytogenes |
Table 9.3: Drugs that inhibit bacterial protein synthesis
Inhibitors of Membrane Function
A small group of antibacterials target the bacterial membrane as their mode of action (table 9.4). The polymyxins are natural polypeptide antibiotics that were first discovered in 1947 as products of Bacillus polymyxa; only polymyxin B and polymyxin E (colistin) have been used clinically. They are lipophilic with detergent-like properties and interact with the lipopolysaccharide component of the outer membrane of gram-negative bacteria, ultimately disrupting both their outer and inner membranes and killing the bacterial cells. Unfortunately, the membrane-targeting mechanism is not a selective toxicity, and these drugs also target and damage the membrane of cells in the kidney and nervous system when administered systemically. Because of these serious side effects and their poor absorption from the digestive tract, polymyxin B is used in over-the-counter topical antibiotic ointments (e.g., Neosporin®).
Oralcolistin was historically used only for bowel decontamination to prevent infections originating from bowel microbes in immunocompromised patients or for those undergoing certain abdominal surgeries. However, the emergence and spread of multidrug-resistant pathogens has led to increased use of intravenous colistin in hospitals, often as a drug of last resort to treat serious infections. The antibacterial daptomycin is a cyclic lipopeptide produced by Streptomyces roseosporus that seems to work like polymyxins, inserting in the bacterial cell membrane and disrupting it. However, in contrast to polymyxin B and colistin, which target only gram-negative bacteria, daptomycin specifically targets gram-positive bacteria. It is typically administered intravenously and seems to be well tolerated, showing reversible toxicity in skeletal muscles.
| Mechanism of Action | Drug Class | Specific Drugs | Spectrum of Activity | Clinical Use |
|---|---|---|---|---|
| Interacts with lipopolysaccharide in the outer membrane of gram-negative bacteria, killing the cell through the eventual disruption of the outer membrane and cytoplasmic membrane | Polymyxins | Polymyxin B | Gram-negative bacteria, including multidrug-resistant strains | Topical preparations to prevent infections in wounds |
| Polymyxin E (colistin) | Gram-negative bacteria, including multidrug-resistant strains | Oral dosing to decontaminate bowels to prevent infections in immunocompromised patients or patients undergoing invasive surgery/procedures. | ||
| Intravenous dosing to treat serious systemic infections caused by multidrug-resistant pathogens | ||||
| Inserts into the cytoplasmic membrane of gram-positive bacteria, disrupting the membrane and killing the cell | Lipopeptide | Daptomycin | Gram-positive bacteria, including multidrug-resistant strains | Complicated skin and skin-structure infections and bacteremia caused by gram-positive pathogens, including MRSA |
Table 9.4: Drugs that inhibit bacterial membrane function
Inhibitors of Nucleic Acid Synthesis
Some antibacterial drugs work by inhibiting nucleic acid synthesis (table 9.5). For example, metronidazole is a semisynthetic member of the nitroimidazole family that is also antiprotozoal. It interferes with DNA replication in target cells. The drug rifampin is a semisynthetic member of the rifamycin family and functions by blocking RNA polymerase activity in bacteria. The RNA polymerase enzymes in bacteria are structurally different from those in eukaryotes, providing selective toxicity against bacterial cells. It is used for the treatment of a variety of infections, but its primary use, often in a cocktail with other antibacterial drugs, is against mycobacteria that cause tuberculosis. Despite the selectivity of its mechanism, rifampin can induce liver enzymes (CYP450 3A4) to increase metabolism of other drugs being administered, leading to hepatotoxicity and negatively influencing the bioavailability and therapeutic effect of the companion drugs.
One member of the quinolone family, a group of synthetic antimicrobials, is nalidixic acid. It was discovered in 1962 as a byproduct during the synthesis of chloroquine, an antimalarial drug. Nalidixic acid selectively inhibits the activity of bacterial DNA gyrase, blocking DNA replication. Chemical modifications to the original quinolone backbone have resulted in the production of fluoroquinolones, like ciprofloxacin and levofloxacin, which also inhibit the activity of DNA gyrase. Ciprofloxacin and levofloxacin are effective against a broad spectrum gram-positive or gram-negative bacterium and are prescribed to treat a wide range of infections, including urinary tract infections, respiratory infections, abdominal infections, and skin infections. However, despite their selective toxicity against DNA gyrase, side effects associated with fluoroquinolones include phototoxicity, neurotoxicity, cardiotoxicity, glucose metabolism dysfunction, and increased risk for tendon rupture.
| Mechanisms of Action | Drug Class | Specific Drugs | Spectrum of activity | Clinical Use |
|---|---|---|---|---|
| Inhibits bacterial RNA polymerase activity and blocks transcription, killing the cell | Rifamycin | Rifampin | Gram-positive and limited gram-negative bacteria , active against Mycobacterium tuberculosis. | Combination therapy for treatment of tuberculosis |
| Inhibits the activity of DNA gyrase and blocks DNA replication, killing the cell | Fluoroquinolones | Ciprofloxacin, ofloxacin, moxifloxacin, levofloxacin | Activity against aerobic, enteric gram-negative bacilli, some are active against Pseudomonas species, selected gram-positive organisms, anaerobes, and mycobacteria. | Wide variety of skin and systemic infections |
Table 9.5: Drugs that inhibit bacterial nucleic acid synthesis
Inhibitors of Metabolic Pathways
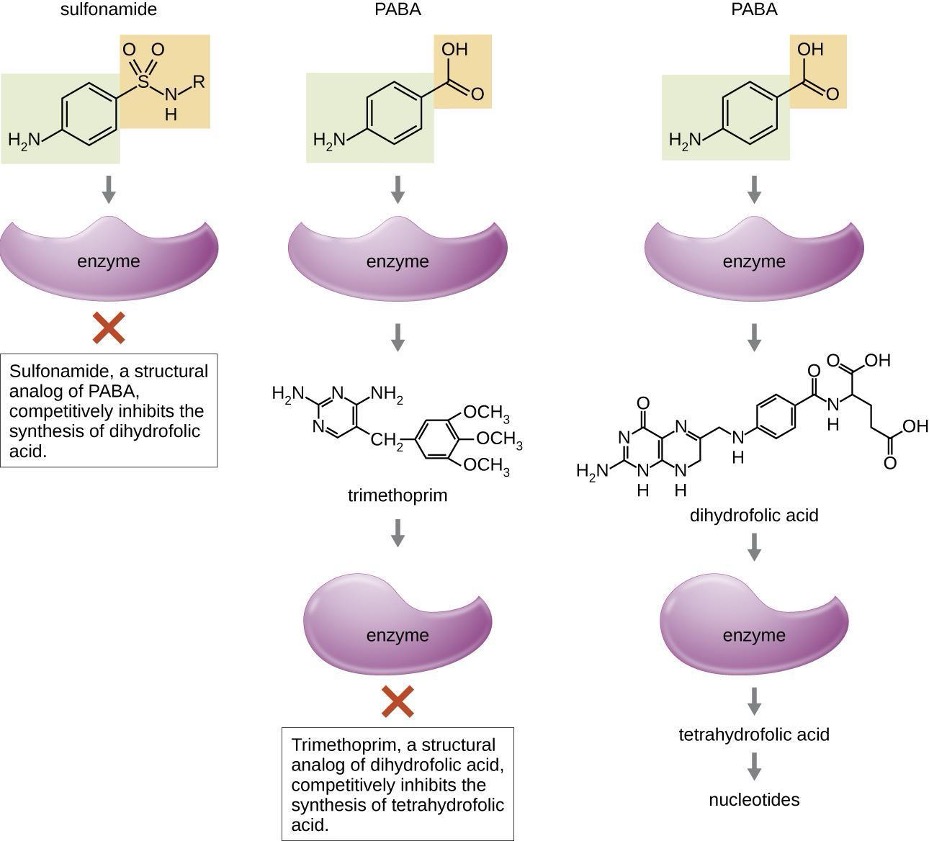
Some synthetic drugs control bacterial infections by functioning as antimetabolites, competitive inhibitors for bacterial metabolic enzymes (table 9.6). The sulfonamides (sulfa drugs) are the oldest synthetic antibacterial agents and are structural analogues of para-aminobenzoic acid (PABA), an early intermediate in folic acid synthesis (figure 9.6). By inhibiting the enzyme involved in the production of dihydrofolic acid, sulfonamides block bacterial biosynthesis of folic acid and, subsequently, pyrimidines and purines required for nucleic acid synthesis. This mechanism of action provides bacteriostatic inhibition of growth against a wide spectrum of gram-positive and gram-negative pathogens. Because humans obtain folic acid from food instead of synthesizing it intracellularly, sulfonamides are selectively toxic for bacteria. However, allergic reactions to sulfa drugs are common. The sulfones are structurally similar to sulfonamides but are not commonly used today except for the treatment of Hansen’s disease (leprosy).
Trimethoprim is a synthetic antimicrobial compound that serves as an antimetabolite within the same folic acid synthesis pathway as sulfonamides. However, trimethoprim is a structural analogue of dihydrofolic acid and inhibits a later step in the metabolic pathway (figure 9.6). Trimethoprim is used in combination with the sulfa drug sulfamethoxazole to treat urinary tract infections, ear infections, and bronchitis. As discussed, the combination of trimethoprim and sulfamethoxazole is an example of antibacterial synergy. When used alone, each antimetabolite only decreases production of folic acid to a level where bacteriostatic inhibition of growth occurs. However, when used in combination, inhibition of both steps in the metabolic pathway decreases folic acid synthesis to a level that is lethal to the bacterial cell. Because of the importance of folic acid during fetal development, sulfa drugs and trimethoprim use should be carefully considered during early pregnancy.
The drug isoniazid is an antimetabolite with specific toxicity for mycobacteria and has long been used in combination with rifampin or streptomycin in the treatment of tuberculosis. It is administered as a prodrug, requiring activation through the action of an intracellular bacterial peroxidase enzyme, forming isoniazid-nicotinamide adenine dinucleotide (NAD) and isoniazid-nicotinamide adenine dinucleotide phosphate (NADP). This ultimately prevents the synthesis of mycolic acid, which is essential for mycobacterial cell walls. Possible side effects of isoniazid use include hepatotoxicity, neurotoxicity, and hematologic toxicity (anemia).
Inhibitor of ATP Synthase
Bedaquiline, representing the synthetic antibacterial class of compounds called the diarylquinolines, uses a novel mode of action that specifically inhibits mycobacterial growth. The drug acts by blocking the ion-binding sites of mycobacterial ATP synthase, resulting in ATP depletion. This damages the energy-producing capacity of mycobacteria, disrupts pH homeostasis, and ultimately inhibits the growth of the strain. Due to its side effects, including hepatotoxicity and potentially lethal heart arrhythmia, its use is reserved for serious, otherwise untreatable cases of tuberculosis.
| Metabolic Pathway Target | Mechanism of Action | Drug Class | Specific Drugs | Spectrum of Activity |
|---|---|---|---|---|
| Folic acid synthesis | Inhibits the enzyme involved in production of dihydrofolic acid | Sulfonamides | Sulfamethoxazole | Gram-positive and gram-negative bacterim |
| Sulfones | Dapsone | Mycobacterium leprae | ||
| Inhibits the enzyme involved in the production of tetrahydrofolic acid | Not applicable | Trimethoprim | Gram-positive and gram-negative bacteria | |
| Mycolic acid synthesis | Interferes with the synthesis of mycolic acid | Not applicable | Isoniazid | Mycobacterium spp., including M. tuberculosis |
Table 9.6: Antimetabolite drugs
| Drugs to consider utilizing for certain bacterial infections | |
|---|---|
| GRAM POSITIVES | |
| MSSA | Oral: cephalexin, cefadroxil, dicloxacillin, amox/clav; IV: cefazolin, oxacillin, nafcillin |
| MRSA | Oral: SMX/TMP, doxycycline, clindamycin, linezolid, tedizolid, omadacycline, delafloxacin; IV: vancomycin, daptomycin, telavancin, dalbavancin, oritavancin, ceftaroline, tigecycline, eravacycline; all oral options also come as IV |
| Enterococci | Ampicillin, then vancomycin; if VRE: linezolid, daptomycin, oritavancin, tigecycline, omadacycline, eravacycline, nitrofurantoin (cystitis only), Fosfomycin (cystitis only) |
| Strep. pyogenes or Strep. agalactiae | Penicillin, amoxicillin, clindamycin |
| Strep. pneumonia or Viridans group Streptococci | Ceftriaxone, levofloxacin, amoxicillin, amoxicillin-clavulanic acid (beware penicillin & macrolide resistance) |
| Listeria monocytogenes | Ampicillin |
| GRAM NEGATIVES | |
| Pseudomonas aeruginosa | Oral: ciprofloxacin, levofloxacin, delafloxacin; IV: pip/taz, ceftazidime, ceftazidime-avibactam, cefepime, ceftolozane-tazobactam, cefiderocol, imipenem-cilastatin +/- relebactam, meropenem, aztreonam, aminoglycosides, polymyxins |
| E. coli, Klebsiella | Oral: cephalexin, amoxicillin-clavulanic acid, SMX/TMP, nitrofurantoin (cystitis only), fosfomycin, ciprofloxacin, levofloxacin, delafloxacin IV: ceftriaxone, ampicillin-sulbactam (beware resistance), cefepime, piperacillin-tazobactam, carbapenems. |
| ESBL-producer / Drug-Resistant GNR | Carbapenems, ceftolozane-tazobactam, ceftazidime- avibactam, meropenem-vaborbactam, imipenem- cilastatin-relebactam, cefiderocol, polymyxins, aminoglycosides, fosfomycin, nitrofurantoin (cystitis only), tigecycline, omadacycline, eravacycline |
| Carbapenem resistant GNR | ESBL-producer drug list minus carbapenem alone |
| NDM producer | Aztreonam + (MERO/VAB or CAZ/AVI), cefiderocol |
| Stenotrophomonas | SMX/TMP, levofloxacin, minocycline |
| Salmonella typhi | Fluoroquinolone, SMX/TMP, ceftriaxone |
Table 9.7: Drugs to consider utilizing for certain bacterial infections
9.3 Mechanisms of Other Antimicrobial Drugs
Because fungi, protozoa, and helminths are eukaryotic, their cells are very similar to human cells, making it more difficult to develop drugs with selective toxicity. Additionally, viruses replicate within human host cells, making it difficult to develop drugs that are selectively toxic to viruses or virus-infected cells. Despite these challenges, there are antimicrobial drugs that target fungi, protozoa, helminths, and viruses, and some even target more than one type of microbe. Table 9.8, table 9.9, table 9.10, and table 9.11 provide examples for antimicrobial drugs in these various classes.
Antifungal Drugs
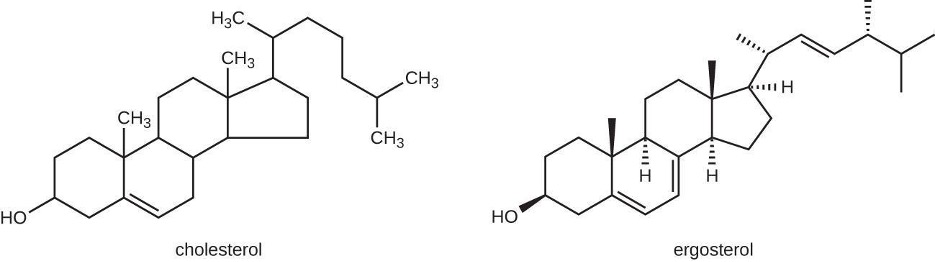
The most common mode of action for antifungal drugs is the disruption of the cell membrane. Antifungals take advantage of small differences between fungi and humans in the biochemical pathways that synthesize sterols. Sterols are important in maintaining proper membrane fluidity and, hence, proper function of the cell membrane. For most fungi, the predominant membrane sterol is ergosterol. Because human cell membranes use cholesterol, instead of ergosterol, antifungal drugs that target ergosterol synthesis are selectively toxic (figure 9.7).
The imidazoles are synthetic fungicides that disrupt ergosterol biosynthesis; they are commonly used in medical applications and also in agriculture to keep seeds and harvested crops from molding. Examples include miconazole, ketoconazole, and clotrimazole, which are used to treat fungal skin infections such as tinea corporis (ringworm), specifically tinea pedis (athlete’s foot), and tinea cruris (jock itch). These infections are commonly caused by dermatophytes of the genera Trichophyton, Epidermophyton, and Microsporum. Miconazole is also used predominantly for the treatment of vaginal yeast infections caused by the fungus Candida, and ketoconazole is used for the treatment of tinea versicolor and dandruff, which both can be caused by the fungus Malassezia.
The triazole drugs, including fluconazole, itraconazole, and voriconazole, also inhibit ergosterol biosynthesis. However, they can be administered orally or intravenously for the treatment of several types of systemic yeast infections, including oral thrush and cryptococcal meningitis, both of which are prevalent in immunocompromised patients. The triazoles also exhibit more selective toxicity, compared with the imidazoles, and are associated with fewer side effects.
The allylamines, a structurally different class of synthetic antifungal drugs, inhibit an earlier step in ergosterol biosynthesis. Examples include naftifine (Naftin®) and terbinafine (Lamisil®) which can be used topically for the treatment of dermatophytic skin infections like athlete’s foot, ringworm, and jock itch. Oral treatment with terbinafine is also used for the treatment of fingernail and toenail fungus, but it can be associated with the rare side effect of hepatotoxicity.
Polyenes are a class of antifungal agents naturally produced by certain actinomycete soil bacteria and are structurally related to macrolides. These large, lipophilic molecules bind to ergosterol in fungal cytoplasmic membranes, thus creating pores. Common examples include nystatin, amphotericin B, and natamycin. Nystatin is typically used as a topical treatment for yeast infections of the skin, mouth, and vagina. The drug amphotericin B is used for systemic fungal infections like aspergillosis, cryptococcal meningitis, histoplasmosis, blastomycosis, and candidiasis. Amphotericin B was the only antifungal drug available for several decades, but its use is associated with some serious side effects, including nephrotoxicity.
Amphotericin B is often used with flucytosine, a fluorinated pyrimidine analog converted by a fungal-specific enzyme into a toxic product that interferes with DNA replication and protein synthesis in fungi. Flucytosine is also associated with hepatotoxicity and bone marrow depression.
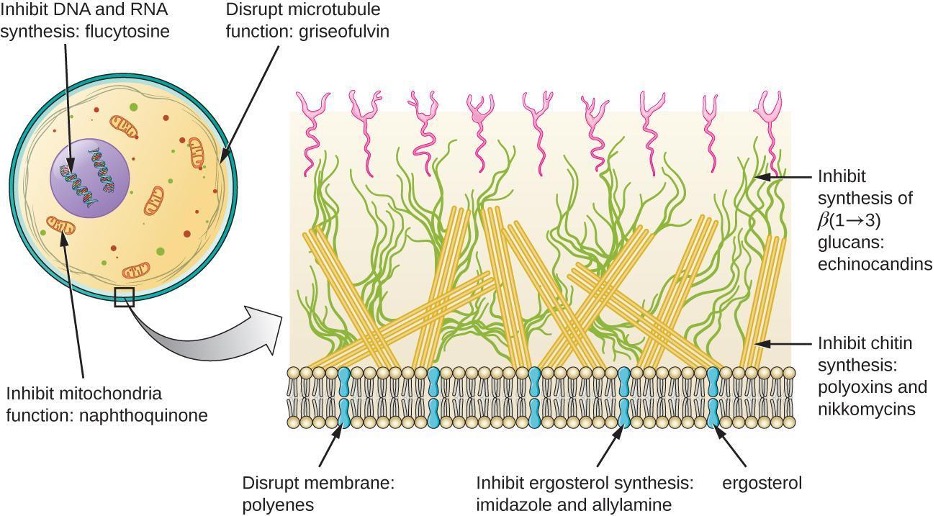
Beyond targeting ergosterol in fungal cell membranes, there are a few antifungal drugs that target other fungal structures (figure 9.8). The echinocandins, (caspofungin, anidulafungin, micafungin, & rezafungin) are a group of naturally produced antifungal compounds that block the synthesis of β (1→3) glucan found in fungal cell walls but not found in human cells. Caspofungin is used for the treatment of invasive aspergillosis as well as candidemia.
The naturally produced antifungal griseofulvin is thought to specifically disrupt fungal cell division by interfering with microtubules involved in spindle formation during mitosis. It was one of the first antifungals, but its use is associated with hepatotoxicity, photosensitivity, and severe skin reactions. It is typically administered orally to treat various types of dermatophytic skin infections when other topical antifungal treatments are ineffective.
There are a few drugs that act as antimetabolites against fungal processes. For example, atovaquone, a representative of the naphthoquinone drug class, is a semisynthetic antimetabolite for fungal and protozoal versions of a mitochondrial cytochrome important in electron transport. Structurally, it is an analog of coenzyme Q, with which it competes for electron binding. It is particularly useful for the treatment of Pneumocystis pneumonia caused by Pneumocystis jirovecii. The antibacterial sulfamethoxazole-trimethoprim combination also acts as an antimetabolite against P. jirovecii.
Table 9.8 shows the various therapeutic classes of antifungal drugs, categorized by mode of action, with examples of each.
| Mechanism of Action | Drug Class | Specific Drugs | Clinical Uses |
|---|---|---|---|
| Inhibit ergosterol synthesis | Imidazoles | miconazole, ketoconazole, clotrimazole | Fungal skin infections and vaginal yeast infections |
| Triazoles | fluconazole, Itraconazole, voriconazole |
Systemic yeast infections, oral thrush, and cryptococcal meningitis | |
| Allylamines | terbinafine | Dermatophytic skin infections (athlete’s foot, ringworm, jock itch), and infections of fingernails and toenails | |
| Bind ergosterol in the cell membrane and create pores that disrupt the membrane | Polyenes | nystatin | Used topically for yeast infections of skin, mouth, and vagina; also used for fungal infections of the intestine |
| amphotericin B | Variety systemic fungal infections | ||
| Inhibit cell wall synthesis | Echinocandins | caspofungin, anidulafungin, micafungin, rezafungin |
Systemic yeast infections |
| Inhibit microtubules and cell division | Not applicable | Griseofulvin | Dermatophytic skin infections |
Table 9.8: Common antifungal drugs
Antiprotozoal Drugs
There are a few mechanisms by which antiprotozoal drugs target infectious protozoans (table 9.10). Some are antimetabolites, such as atovaquone, proguanil, and artesunate. Atovaquone, in addition to being antifungal, blocks electron transport in protozoans and is used for treating protozoan infections including malaria, babesiosis, and toxoplasmosis. Proguanil is another synthetic antimetabolite that is processed in parasitic cells into its active form, which inhibits protozoan folic acid synthesis. It is often used in combination with atovaquone, and the combination is marketed as Malarone® for both malaria treatment and prevention.
Artesunate, an artemisinin derivative, is a semisynthetic derivative of artemisinin that is more water soluble than the natural version, which makes it more bioavailable for severe malaria. Artesunate is a prodrug that is metabolized to its active form, dihydroartemisinic (DHA). DHA contains an endoperoxide bridge activated by heme iron binding, resulting in oxidative stress, inhibition of protein and nucleic acid synthesis, thereby decreasing parasite growth and survival. Due to the rise in resistance to antimalarial drugs, artemisinins are used in combination with other antimalarial compounds in artemisinin-based combination therapy (ACT).
Several antimetabolites are used for the treatment of toxoplasmosis caused by the parasite Toxoplasma gondii. The synthetic sulfa drug sulfadiazine competitively inhibits the enzyme para-aminobenzoic acid (PABA) inhibiting folic acid synthesis and can be used to treat toxoplasmosis. Pyrimethamine is a synthetic drug that inhibits the enzyme dihydrofolate reductase resulting in inhibition of the folic acid production pathway. Side effects of pyrimethamine include decreased bone marrow activity that may cause increased bruising and low red blood cell counts. Two classes of antiprotozoal drugs interfere with nucleic acid synthesis: nitroimidazoles and quinolines.
Nitroimidazoles, including synthetic tinidazole and semisynthetic metronidazole, which was discussed previously as an antibacterial drug, are useful in combating a wide variety of protozoan pathogens, such as Giardia lamblia, Entamoeba histolytica, and Trichomonas vaginalis. Upon introduction into these cells in low-oxygen environments, nitroimidazoles become activated and introduce DNA strand breakage, interfering with DNA replication in target cells. Unfortunately, carcinogenicity has been seen in mice and rats treated chronically with metronidazole, and it is recommended that unnecessary use be avoided.
The aminoquinolines such as chloroquine (CQ) are a class of synthetic compounds related to quinine, which has a long history of use against malaria. CQ increases pH and accumulates in the food vacuole of parasites leading to the release of toxic products including hematin. CQ inhibits the polymerization and detoxification of hematin and interferes with the degradation of host erythrocyte hemoglobin, leading to the accumulation of free hematin that is highly toxic to Plasmodium, resulting in dissolution of the cell membrane and, ultimately, death of the parasites. Additionally, CQ can insert into the DNA double helix structure of Plasmodium impacting DNA replication and RNA transcription, to inhibit growth and reproduction of Plasmodium.[4]
Long-term prophylactic use of chloroquine may result in serious side effects, including hallucinations or cardiac issues. Patients with glucose-6-phosphate dehydrogenase deficiency experience severe anemia when treated with chloroquine.
| Mechanism of Action | Drug Class | Specific Drugs | Clinical Uses |
|---|---|---|---|
| Inhibit electron transport in mitochondria | Naphthoquinone | Atovaquone | Malaria, babesiosis, and toxoplasmosis |
| Inhibit folic acid synthesis | Not applicable | Proguanil | Combination therapy with atovaquone for malaria treatment and prevention |
| Sulfonamide | Sulfadiazine | Toxoplasmosis | |
| Not applicable | Pyrimethamine | Treatment of toxoplasmosis when used in combination with sulfonamide | |
| Produces damaging reactive oxygen species | Not applicable | Artemisinin | Combination therapy to treat malaria |
| Inhibit DNA synthesis | Nitroimidazoles | Metronidazole, tinidazole | Infections caused by Giardia lamblia, Entamoeba histolytica, and Trichomonas vaginalis |
| Inhibit heme detoxification | Aminoquinolines | Chloroquine | Malaria |
Table 9.9: Common antiprotozoal drugs
Antihelminthic Drugs
Because helminths are multicellular eukaryotes like humans, developing drugs with selective toxicity against them is extremely challenging. Despite this, several effective classes have been developed (table 9.10). Synthetic benzimidazoles, like mebendazole and albendazole, bind to helminthic β-tubulin, preventing microtubule formation. Microtubules in the intestinal cells of the worms seem to be particularly affected, leading to a reduction in glucose uptake. Besides their activity against a broad range of helminths, benzimidazoles are also active against many protozoans, fungi, and viruses. Their use for inhibiting mitosis and cell cycle progression in cancer cells is also under study.[5] Possible side effects of their use include gastrointestinal (abdominal pain, anorexia, diarrhea, nausea, vomiting) and bone marrow suppression (when used at higher doses or for prolonged duration).
The avermectins are members of the macrolide family that were first discovered from a Japanese soil isolate, Streptomyces avermectinius. A more potent semisynthetic derivative of avermectin is ivermectin, which binds to glutamate-gated chloride channels specific to invertebrates including helminths, blocking neuronal transmission and causing starvation, paralysis, and death of the worms. Ivermectin is used to treat roundworm diseases, including onchocerciasis (also called river blindness, caused by the worm Onchocerca volvulus) and strongyloidiasis (caused by the worm Strongyloides stercoralis or S. fuelleborni). Ivermectin can also treat parasitic insects like mites, lice, and bed bugs, and is nontoxic to humans.[6][7][8]
Another synthetic antihelminthic drug is praziquantel which is particularly useful for the treatment of schistosomiasis (caused by blood flukes from three genera of Schistosoma). Its mode of action is binding to a TRPM2-like channel causing the influx of calcium into the worm, resulting in intense spasm and paralysis of the worm.[9]
| Mechanism of Action | Drug Class | Specific Drugs | Clinic Uses |
|---|---|---|---|
| Inhibit microtubule formation, reducing glucose uptake | Benzimidazoles | Mebendazole, albendazole | Ancylostoma duodenale or Necator americanus (hookworms), Ascaris lumbricoides (roundworms), Enterobius vermicularis (pinworms), and Trichuris trichiura (whipworms) |
| Block neuronal transmission, causing paralysis and starvation | Avermectins | Ivermectin | Onchocerciasis (river blindness) due to the immature form of Onchocerca volvulus. intestinal (e.g., non-disseminated) strongyloidiasis due to Strongyloides stercoralis. |
| Induce calcium influx | Not applicable | Praziquantel | Schistosomiasis (blood flukes) |
Table 9.10: Common antihelminthic drugs
Antiviral Drugs
Unlike the complex structure of fungi, protozoa, and helminths, viruses have a simpler composition consisting of nucleic acid, a protein coat, viral enzymes, and, sometimes, a lipid envelope. Furthermore, viruses are obligate intracellular pathogens that use the host’s cellular machinery to replicate. These characteristics make it difficult to develop drugs with selective toxicity against viruses.
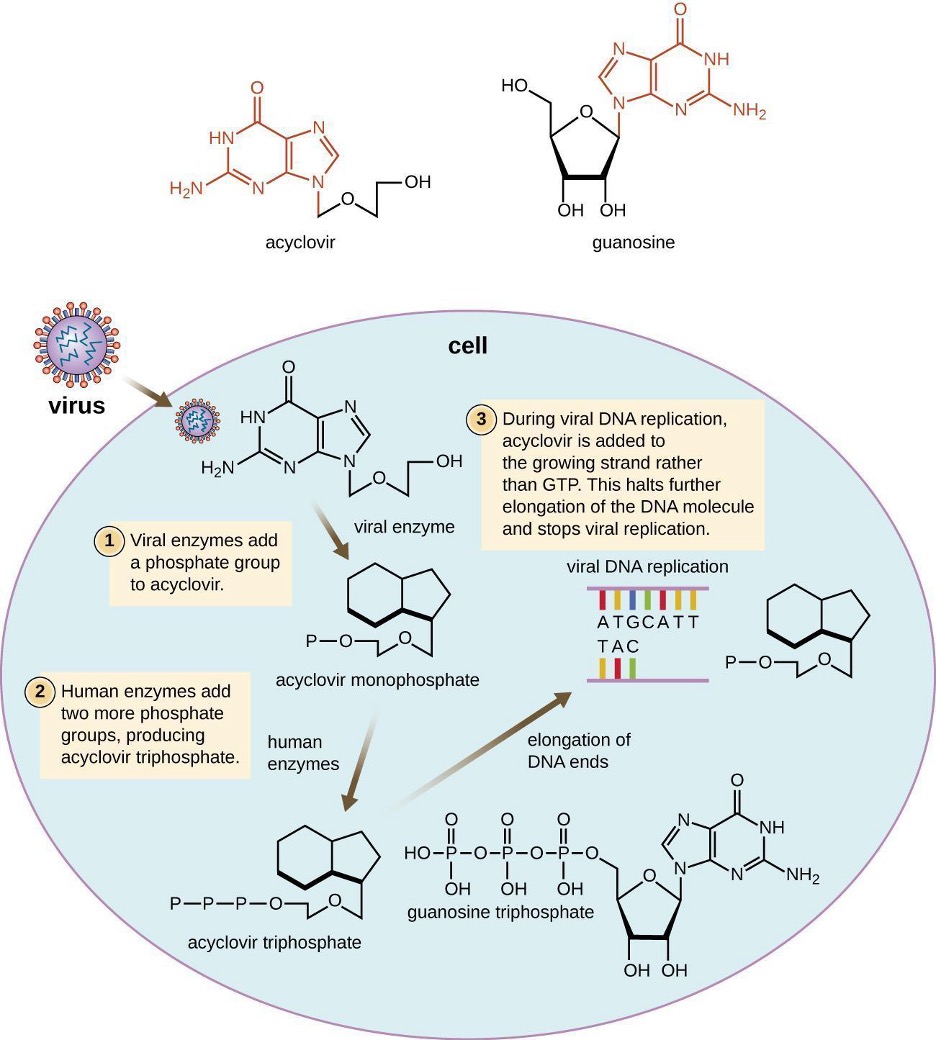
Many antiviral drugs are nucleoside analogs and function by inhibiting nucleic acid biosynthesis. For example, acyclovir (marketed as Zovirax®) is a synthetic analog of the nucleoside guanosine (figure 9.9). It is activated by virus-specific thymidine kinase and then further converted to acyclovir triphosphate by other cellular enzymes. Acyclovir triphosphate inhibits DNA synthesis and viral replication by competing with deoxyguanosine triphosphate for viral DNA polymerase and being incorporated into viral DNA. Acyclovir and its derivatives are frequently used to treat herpes virus infections, including genital herpes, chickenpox, and shingles infections. Acyclovir can be administered either topically or systemically, depending on the infection. One possible side effect of its use includes nephrotoxicity.
Ribavirin, another synthetic guanosine analog, works by a mechanism of action that is not entirely clear. It appears to interfere with both DNA and RNA synthesis, perhaps by reducing intracellular pools of guanosine triphosphate (GTP). Ribavirin also appears to inhibit the RNA polymerase of hepatitis C virus. It is primarily used for the treatment of the RNA viruses like hepatitis C (in combination therapy with interferon) and respiratory syncytial virus. Possible side effects of ribavirin use include anemia and developmental effects on unborn children in pregnant patients. In recent years, another nucleotide analog, sofosbuvir (Solvaldi®), has also been developed for the treatment of hepatitis C. Sofosbuvir is a uridine analog that interferes with viral polymerase activity. It is commonly co-administered with ribavirin, with and without interferon.
Inhibition of nucleic acid synthesis is not the only target of synthetic antivirals. Although the mode of action of amantadine and its relative rimantadine are not entirely clear, these drugs appear to bind to a transmembrane protein that is involved in the escape of the influenza virus from endosomes. Blocking escape of the virus also prevents viral RNA release into host cells and subsequent viral replication. Increasing resistance has limited the use of amantadine and rimantadine in the treatment of influenza A. Use of amantadine can result in neurological side effects, but the side effects of rimantadine seem less severe. Interestingly, because of their effects on brain chemicals such as dopamine and NMDA (N-methyl D-aspartate), amantadine and rimantadine are also used for the treatment of Parkinson’s disease.
Neuraminidase inhibitors, including oseltamivir (Tamiflu®) and zanamivir (Relenza®), specifically target influenza viruses by blocking the activity of influenza virus neuraminidase, preventing the release of the virus from infected cells. These three antivirals can decrease flu symptoms and shorten the duration of illness, but they differ in their modes of administration: oseltamivir is administered orally and zanamivir is inhaled. Resistance to these neuraminidase inhibitors still seems to be minimal.
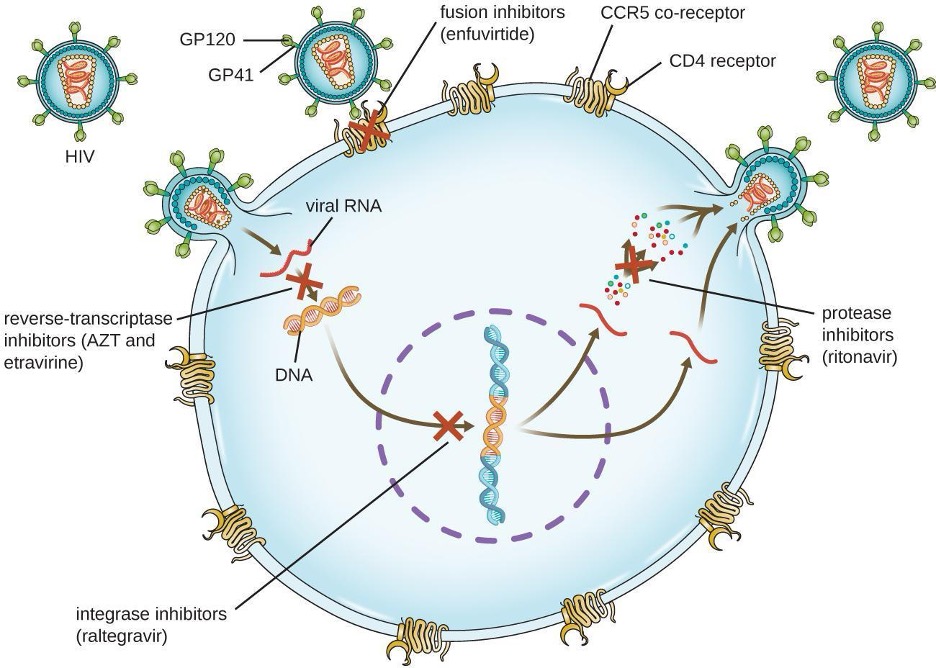
Viruses with complex life cycles, such as HIV, can be more difficult to treat. First, HIV targets CD4-positive white blood cells, which are necessary for a normal immune response to infection. Second, HIV is a retrovirus, meaning that it converts its RNA genome into a DNA copy that integrates into the host cell’s genome, thus hiding within host cell DNA. Third, the HIV reverse transcriptase lacks proofreading activity and introduces mutations that allow for rapid development of antiviral drug resistance. To help prevent the emergence of resistance, a combination of specific synthetic antiviral drugs is typically used in ART for HIV (figure 9.10). Table 9.11 shows the various therapeutic classes of antiviral drugs, categorized by mode of action, with examples of each.
Targeted therapy for HIV
Fusion/entry inhibitors are a class of drugs employed in HIV treatment that specifically target the initial stages of the virus life cycle blocking the fusion of HIV with the host cell membrane preventing viral entry into host cells (figure 9.11).
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are a class of antiretroviral drugs used in the treatment of HIV. They work by binding to the viral enzyme reverse transcriptase, disrupting its function, preventing the conversion of viral RNA into DNA, thereby hindering the replication of the virus.
Nucleoside reverse transcriptase inhibitors (NRTIs) act by mimicking the building blocks of DNA. Once incorporated into the growing viral DNA chain, NRTIs terminate the process, preventing further replication and inhibiting the progression of HIV infection.
Integrase inhibitors are a class of antiretroviral drugs that specifically target the viral enzyme integrase. By blocking the integration of viral DNA into the host cell genome, these inhibitors disrupt the virus’s ability to establish a permanent infection, ultimately suppressing HIV replication.
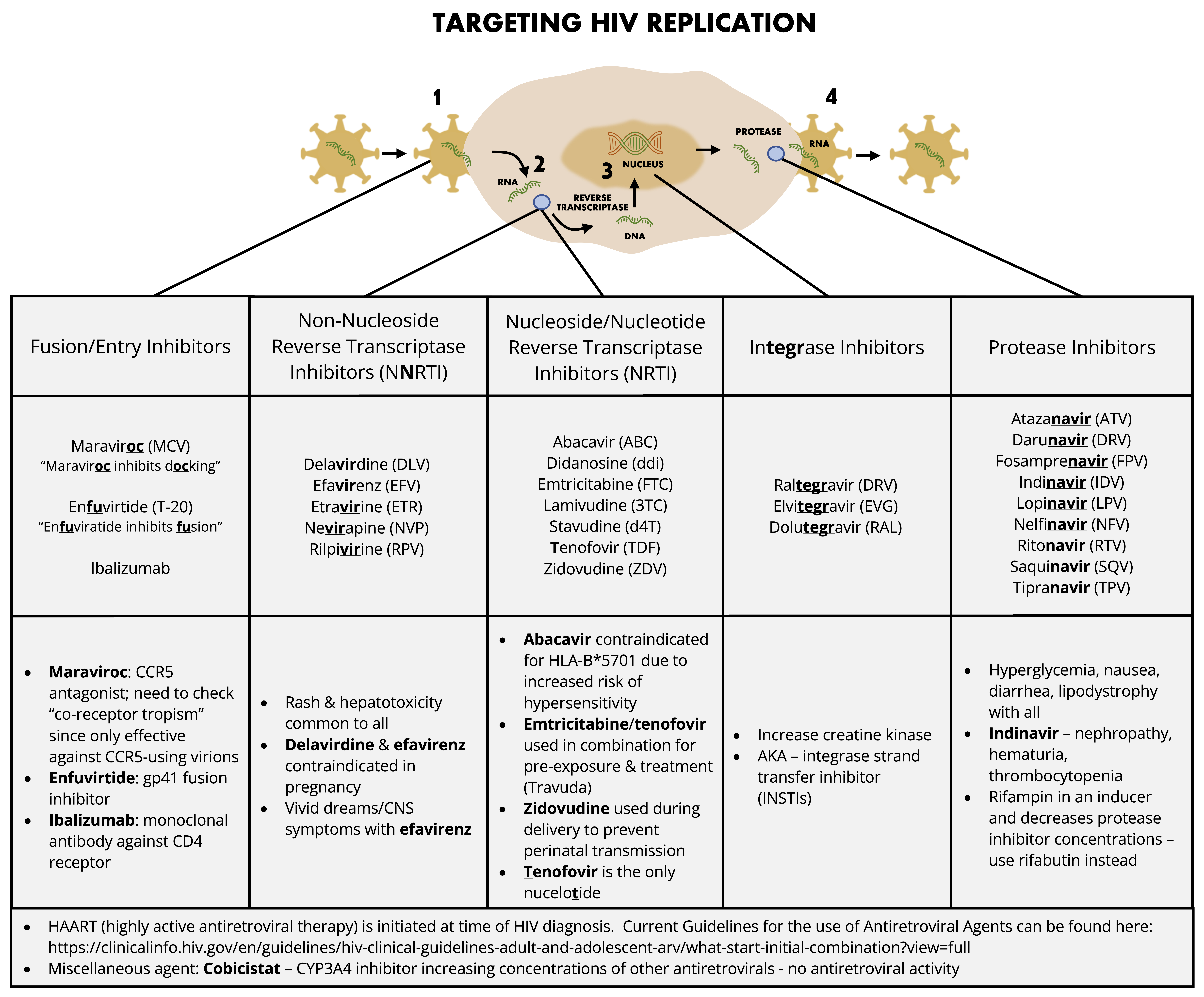
Protease inhibitors are designed to block the activity of the viral enzyme protease. By inhibiting protease, these drugs prevent the maturation of newly formed viral particles, ultimately curtailing the production of infectious HIV and slowing the progression of the disease.
Targeted therapy for Hepatitis C
Glecaprevir and pibrentasvir (Mavyret®) are antiviral drugs used in combination for the treatment of chronic hepatitis C infections. Glecaprevir is an NS3/4A protease inhibitor, while pibrentasvir is an NS5A inhibitor. Together, they work synergistically to disrupt the viral life cycle, inhibiting viral replication and assembly. The mechanism of action involves glecaprevir inhibiting the NS3/4A protease, a key enzyme in the viral polyprotein processing, and pibrentasvir targeting the NS5A protein, crucial for viral RNA replication and assembly. This dual mechanism results in a high barrier to resistance and improved efficacy against a broad range of hepatitis C virus genotypes. Common adverse effects of the glecaprevir/pibrentasvir combination therapy include fatigue, headache, and nausea.
| Mechanism of Action | Drug | Clinical Uses |
|---|---|---|
| Nucleoside analog inhibition of nucleic acid synthesis | Acyclovir | Herpes virus infections |
| zidovudine | HIV infections | |
| Ribavirin | Hepatitis C virus and respiratory syncytial virus infections | |
| Non-nucleoside noncompetitive inhibition | Etravirine | HIV infections |
| Inhibit escape of virus from endosomes | Amantadine, rimantadine | Infections with influenza virus |
| Inhibit neuraminidase | Oseltamivir, zanamivir | Infections with influenza virus |
| Inhibition of protease | Ritonavir | HIV infections |
| Inhibition of integrase | Raltegravir | HIV infections |
| Inhibition of membrane fusion | Enfuvirtide | HIV infections |
Table 9.11: Antiviral drugs
9.4 Drug Resistance
Antimicrobial resistance is not a new phenomenon. In nature, microbes are constantly evolving to overcome the antimicrobial compounds produced by other microorganisms. Human development of antimicrobial drugs and their widespread clinical use has simply provided another selective pressure that promotes further evolution. Several important factors can accelerate the evolution of drug resistance. These include the overuse and misuse of antimicrobials, inappropriate use of antimicrobials, subtherapeutic dosing, and patient noncompliance with the recommended course of treatment.
Some organisms, such as Pseudomonas with porin and permeability changes, are more likely to have the intrinsic resistance machinery necessary to have reduced susceptibility to certain agents. Other organisms expressing resistance to antimicrobials may be more likely due to acquired resistance mechanisms. Many genes responsible for drug resistance are found on plasmids or in transposons that can be transferred easily between microbes through horizontal gene transfer. Transposons can also move resistance genes between plasmids and chromosomes to promote the spread of resistance. E. coli is a bacterial example that has acquired genes for ESBL production via plasmid exchange.
Mechanisms for Drug Resistance
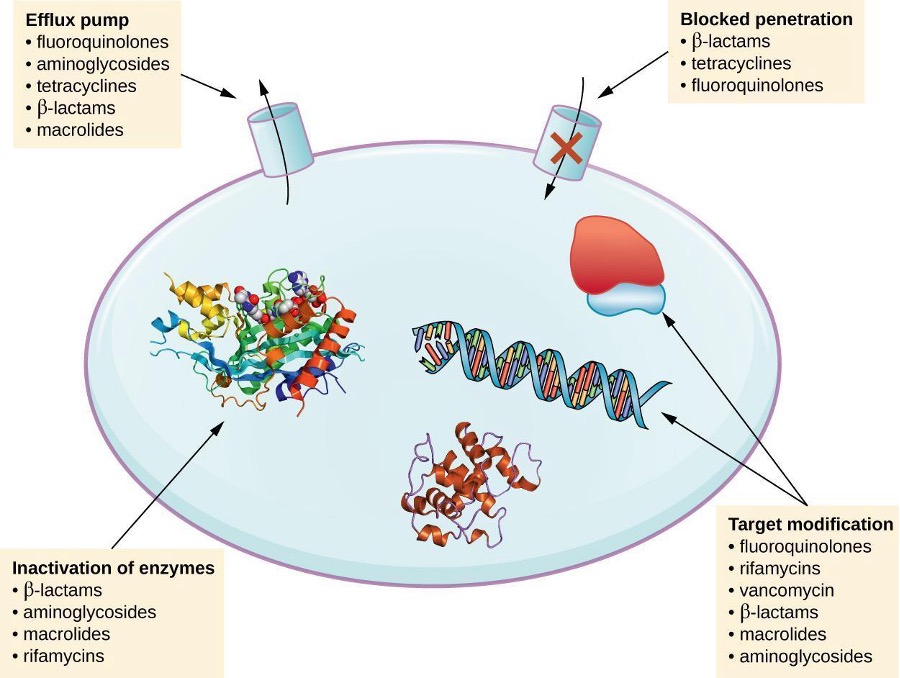
There are several common mechanisms for drug resistance, which are summarized in figure 9.12. These mechanisms include enzymatic modification of the drug, modification of the antimicrobial target, and prevention of drug penetration or accumulation.
Drug Modification or Inactivation
Resistance genes may code for enzymes that chemically modify an antimicrobial, thereby inactivating it, or destroy an antimicrobial through hydrolysis. Resistance to many types of antimicrobials occurs through this mechanism. For example, aminoglycoside resistance can occur through enzymatic transfer of chemical groups to the drug molecule, impairing the binding of the drug to its bacterial target. For β-lactams, bacterial resistance can involve the enzymatic hydrolysis of the β-lactam bond within the β-lactam ring of the drug molecule. Once the β-lactam bond is broken, the drug loses its antibacterial activity. This mechanism of resistance is mediated by β-lactamases, which are the most common mechanism of β-lactam resistance. Inactivation of rifampin commonly occurs through glycosylation, phosphorylation, or adenosine diphosphate (ADP) ribosylation, and resistance to macrolides and lincosamides can also occur due to enzymatic inactivation of the drug or modification.
Prevention of Cellular Uptake or Efflux
Microbes may develop resistance mechanisms that involve inhibiting the accumulation of an antimicrobial drug, which then prevents the drug from reaching its cellular target. This strategy is common among gram-negative pathogens and can involve changes in outer membrane lipid composition, porin channel selectivity, and/or porin channel concentrations. For example, a common mechanism of carbapenem resistance among Pseudomonas aeruginosa is to decrease the amount of its OprD porin, which is the primary portal of entry for carbapenems through the outer membrane of this pathogen. Additionally, many gram-positive and gram-negative pathogenic bacteria produce efflux pumps that actively transport an antimicrobial drug out of the cell and prevent the accumulation of drug to a level that would be antibacterial. For example, resistance to β-lactams, tetracyclines, and fluoroquinolones commonly occurs through active efflux out of the cell, and it is rather common for a single efflux pump to have the ability to translocate multiple types of antimicrobials.
Target Modification
Because antimicrobial drugs have very specific targets, structural changes to those targets can prevent drug binding, rendering the drug ineffective. Through spontaneous mutations in the genes encoding antibacterial drug targets, bacteria have an evolutionary advantage that allows them to develop resistance to drugs. This mechanism of resistance development is quite common. Genetic changes impacting the active site of penicillin-binding proteins (PBPs) can inhibit the binding of β-lactam drugs and provide resistance to multiple drugs within this class. This mechanism is very common among strains of Streptococcus pneumoniae, which alter their own PBPs through genetic mechanisms. In contrast, strains of Staphylococcus aureus develop resistance to methicillin (MRSA) through the acquisition of a new low-affinity PBP, rather than structurally altering their existing PBPs. Not only does this new low-affinity PBP provide resistance to methicillin, but it provides resistance to virtually all β-lactam drugs, with the exception of the newer fifth generation cephalosporins designed specifically to kill MRSA. Other examples of this resistance strategy include alterations in
- ribosome subunits, providing resistance to macrolides, tetracyclines, and aminoglycosides;
- lipopolysaccharide (LPS) structure, providing resistance to polymyxins;
- RNA polymerase, providing resistance to rifampin;
- DNA gyrase, providing resistance to fluoroquinolones;
- metabolic enzymes, providing resistance to sulfa drugs, sulfones, and trimethoprim; and
- peptidoglycan subunit peptide chains, providing resistance to glycopeptides.
Target Overproduction or Enzymatic Bypass
When an antimicrobial drug functions as an antimetabolite, targeting a specific enzyme to inhibit its activity, there are additional ways that microbial resistance may occur. First, the microbe may overproduce the target enzyme such that there is enough antimicrobial-free enzyme to do the proper enzymatic reaction. Second, the bacterial cell may develop a bypass that circumvents the need for the functional target enzyme. Both strategies have been found as mechanisms of sulfonamide resistance. Vancomycin resistance among S. aureus has been shown to involve the decreased cross-linkage of peptide chains in the bacterial cell wall, which provides an increase in targets for vancomycin to bind to in the outer cell wall. Increased binding of vancomycin in the outer cell wall provides a blockage that prevents free drug molecules from penetrating to where they can block new cell wall synthesis.
Target Mimicry
A recently discovered mechanism of resistance called target mimicry involves the production of proteins that prevent drugs from binding to their bacterial cellular targets. For example, fluoroquinolone resistance by Mycobacterium tuberculosis can involve producing a protein that resembles DNA. This protein is called MfpA (Mycobacterium fluoroquinolone resistance protein A). The mimicry of DNA by MfpA results in DNA gyrase binding to MfpA, preventing the binding of fluoroquinolones to DNA gyrase.
Multidrug-Resistant Microbes and Cross Resistance
From a clinical perspective, our greatest concerns are multidrug-resistant microbes (MDRs) and cross resistance. MDRs are colloquially known as “superbugs” and carry one or more resistance mechanism(s), making them resistant to multiple antimicrobials. In cross-resistance, a single resistance mechanism confers resistance to multiple antimicrobial drugs. For example, having an efflux pump that can export multiple antimicrobial drugs is a common way for microbes to be resistant to multiple drugs by using a single resistance mechanism. In recent years, several clinically important superbugs have emerged, and the CDC reports that superbugs are responsible for more than 2 million infections in the US annually, resulting in at least 23,000 fatalities.[10] Several of the superbugs have been dubbed the ESKAPE pathogens. This acronym refers to the names of the pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) but it is also fitting in that these pathogens are able to “escape” many conventional forms of antimicrobial therapy. As such, infections by ESKAPE pathogens can be difficult to treat and cause many nosocomial infections.
Methicillin-Resistant Staphylococcus aureus (MRSA)
Methicillin, a semisynthetic penicillin, was designed to resist inactivation by β-lactamases. Unfortunately, soon after the introduction of methicillin to clinical practice, methicillin-resistant strains of S. aureus appeared and started to spread. The mechanism of resistance, acquisition of a new low-affinity PBP, provided S. aureus with resistance to all available β-lactams. Strains of methicillin-resistant S. aureus (MRSA) are widespread opportunistic pathogens and a particular concern for skin and other wound infections. These strains may also cause pneumonia and septicemia. Although originally a problem in healthcare settings (hospital-acquired MRSA [HA-MRSA]), MRSA infections are now also acquired through contact with contaminated members of the general public, called community-associated MRSA (CA-MRSA). Approximately one-third of the population carries S. aureus as a member of their normal nasal microbiota without illness, and about 6% of these strains are methicillin resistant.[11][12]
Vancomycin-Resistant Enterococci and Staphylococcus aureus
Vancomycin is only effective against gram-positive organisms, and it is used to treat wound infections, septic infections, endocarditis, and meningitis that are caused by pathogens resistant to other antibiotics. It is considered one of the last lines of defense against such resistant infections, including MRSA. With the rise of antibiotic resistance in the 1970s and 1980s, vancomycin use increased, and it is not surprising that we saw the emergence and spread of vancomycin-resistant enterococci (VRE), vancomycin-resistant S. aureus (VRSA), and vancomycin-intermediate S. aureus (VISA). The mechanism of vancomycin resistance among enterococci is target modification involving a structural change to the peptide component of the peptidoglycan subunits, preventing vancomycin from binding. These strains are typically spread among patients in clinical settings by contact with healthcare workers and contaminated surfaces and medical equipment.
VISA and VRSA strains differ from each other in the mechanism of resistance and the degree of resistance each mechanism confers. VISA strains exhibit intermediate resistance, with a minimum inhibitory concentration (MIC) of 4–8 μg/mL, and the mechanism involves an increase in vancomycin targets. VISA strains decrease the crosslinking of peptide chains in the cell wall, providing an increase in vancomycin targets that trap vancomycin in the outer cell wall. In contrast, VRSA strains acquire vancomycin resistance through horizontal transfer of resistance genes from VRE, an opportunity provided in individuals co-infected with both VRE and MRSA. VRSA exhibits a higher level of resistance, with MICs of 16 μg/mL or higher.[13] In the case of all three types of vancomycin-resistant bacteria, rapid clinical identification is necessary so proper procedures to limit spread can be implemented. Oxazolidinones, like linezolid, are useful for the treatment of these vancomycin-resistant, opportunistic pathogens, as well as MRSA.
Extended-Spectrum β-Lactamase–Producing Gram-Negative Pathogens
Gram-negative pathogens that produce extended-spectrum β-lactamases (ESBLs) show resistance well beyond just penicillins. The spectrum of β-lactams inactivated by ESBLs provides for resistance to all penicillins, cephalosporins, monobactams, and the β-lactamase-inhibitor combinations, but not the carbapenems. An even greater concern is that the genes encoding for ESBLs are usually found on mobile plasmids that also contain genes for resistance to other drug classes (e.g., fluoroquinolones, aminoglycosides, tetracyclines), and may be readily spread to other bacteria by horizontal gene transfer. These multidrug-resistant bacteria are members of the intestinal microbiota of some individuals, but they are also important causes of opportunistic infections in hospitalized patients, from whom they can be spread to other people.
Carbapenem-Resistant Gram-Negative Bacteria
The occurrence of carbapenem-resistant Enterobacterales (CRE) and carbapenem resistance among other gram-negative bacteria (e.g., P. aeruginosa, Acinetobacter baumannii, Stenotrophomonas maltophila) is a growing healthcare concern. These pathogens develop resistance to carbapenems through a variety of mechanisms, including production of carbapenemases (broad-spectrum β-lactamases that inactivate all β-lactams, including carbapenems), active efflux of carbapenems out of the cell, and/or prevention of carbapenem entry through porin channels. Similar to concerns with ESBLs, carbapenem-resistant, gram-negative pathogens are usually resistant to multiple classes of antibacterials, and some have even developed pan-resistance (resistance to all available antibacterials). Infections with carbapenem-resistant, gram-negative pathogens commonly occur in healthcare settings through interaction with contaminated individuals or medical devices, or as a result of surgery.
Multidrug-Resistant Mycobacterium tuberculosis
The emergence of multidrug-resistant Mycobacterium tuberculosis (MDR-TB) and extensively drug-resistant Mycobacterium tuberculosis (XDR-TB) is also of significant global concern. MDR-TB strains are resistant to both rifampin and isoniazid, the drug combination typically prescribed for treatment of tuberculosis. XDR-TB strains are additionally resistant to any fluoroquinolone and at least one of three other drugs (amikacin, kanamycin, or capreomycin) used as a second line of treatment, leaving these patients very few treatment options. Both types of pathogens are particularly problematic in immunocompromised persons, including those suffering from HIV infection. The development of resistance in these strains often results from the incorrect use of antimicrobials for tuberculosis treatment, selecting for resistance.
Figure Descriptions
Figure 9.1: Diagram of process of superinfection. 1: Normal microbiota keeps opportunistic pathogens in check. The image shows many different bacteria, only 1 of which is labeled pathogen. 2: Broad-spectrum antibiotics kill nonresistant cells. The image shows all cells but the pathogen being killed. 3: Drug-resistant pathogens proliferate and can cause a superinfection. The image shows many of the pathogens.
Figure 9.2: Graph with time on the X axis and Plasma Concentration of Drug on the Y axis. IV route increases plasma concentration very quickly and then tapes off. The intramuscular route and oral route increase concentrations more slowly, with the intramuscular route being a bit faster than oral but also dropping off more quickly.
Figure 9.3: An illustration of a cell is shown with a view inside. The double helix is visible in the center, and a label points to it indicating DNA synthesis, fluoroquinolones, ciprofloxacin, levofloxacin, moxifloxacin, RNA synthesis, Rifamycins, and rifampin. Another label points to the cell wall and indicates beta-lactams, penicillins, cephalosporins, monobactams, carbapenems, glycopeptides, vancomycin, and bacitracin. A third label points to the plasma membrane and indicates polymyxins, polymyxin B, colistin, lipopeptide, and daptomycin. Within the cytoplasm, another label points to ribosomes, which include 30s subunit, aminoglycosides, tetracyclines, 50s subunit, macrolides, lincosamides, chloramphenicol, and oxazolidinones. The final label points to the metabolic pathways and indicates folic acid synthesis, sulfonamides, sulfones, trimethoprim, mycolic acid synthesis, and isoniazid.
Figure 9.4: The top of the image shows diagrams of various antibiotics. All have a beta-lactam ring, which is a square made of 3 carbons and nitrogen; one of the carbons has a double-bonded O. The antibiotics shown are penicillin, cephalosporin, monobactam, and carbapenem. Below is a table with the rows: R group, Drug name, spectrum of activity, and route of administration. Penicillin G has an R group of carbon linked to a 6-carbon ring; it is active on G+ and a few G- cells and has a parenteral route of administration. Penicillin V has an R group of a carbon linked to an oxygen linked to a carbon ring. IT affects G+ and a few G- and is administered orally. Ampicillin has an R group of Carbon attached to both an amino group and a carbon ring. It is effective against G+ and more G- than penicillin. It is administered orally and parenterally. Amoxicillin has an R group similar to ampicillin, but the carbon ring has an additional OH. It has similar activity to ampicillin and is administered orally (better than ampicillin). Methicillin has an R group of carbon right with 2 CH3O attached to the ring. It is effective against G+ only, including B-lactam producers. It is administered parenterally.
Figure 9.5: Major classes of protein synthesis-inhibiting antibacterials. Chloramphenicol, macrolides, and lincosamides: bind to the 50S ribosomal subunit, preventing peptide bond formation, and stop protein synthesis. Aminoglycosides: bind to the 30S ribosomal subunit, implant proofreading, resulting in production of faulty proteins. Tetracyclines: bind to the 30S ribosomal subunit, block the binding of tRNAs, thereby inhibiting protein synthesis.
Figure 9.6: PABA binds to an enzyme to produce dihydrofolic acid, which binds to another enzyme to produce tetrahydrofolic acid and nucleotides. Trimethoprim, a structural analog of dihydrofolic acid, completely inhibits the synthesis of tetrahydrofolic acid. Sulfonamide, a structural analog of PABA, competitively inhibits the synthesis of dihydrofolic acid.
Figure 9.7: Cholesterol and ergosterol both have 4 fused carbon rings with a chain of carbons off the top ring. The differences are the placements of a few double bonds.
Figure 9.8: Targets of antifungal drugs: Inhibits mitochondria function: naphthoquinone. Disrupt membrane: polyenes. Inhibit ergosterol synthesis: imidazole and allylamine. Inhibit synthesis of beta(1-3) glucans: echinocandins. Inhibit chitin synthesis: polyoxins and nikko mycins.
Figure 9.9: Acyclovir looks similar to guanosine except that the sugar is replaced with a short carbon chain. Step 1: Viral enzyme adds a phosphate group to acyclovir. Step 2: Human enzymes add two more phosphate groups, producing acyclovir triphosphate. Step 3: During viral DNA replication, acyclovir is added to the growing strand rather than GTP. This halts further elongation of the DNA molecule and stops viral replication.
Figure 9.10: Diagram showing HIV infection and locations where drugs can stop the infection. GP120 and G(42 are proteins that are on the surface of the virus and bind to CD4 receptor and CCR5. Enfuvirtide is a fusion inhibitor that blocks this process. When the virus enters, it produces DNA from RNA, this can be blocked by AZT and etravirine which are reverse-transcriptase inhibitors. Next, the viral DNA integrates into the host DNA. Raltegravir is an integrase inhibitor and blocks this step. Finally the virus is rebuilt. Ritonavir is a protease inhibitor and blocks this step.
Figure 9.11: An intricate diagram detailing the targeted therapy for HIV is represented by a series of grey columns arranged in an accompanying arrow table. The descriptions of the columns are given left to right. Starting leftmost, an arrow points to RNA contained within a vesicle, indicating the location of Fusion/Entry Inhibitors. Within this section, Maraviroc (MCV) is described as inhibiting docking, Enfuvirtide (T-20) inhibits fusion, and Ibalizumab is mentioned as a monoclonal antibody against the CD4 receptor. Next, an arrow extends towards RNA after its entry into the cell, representing Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI). The drugs listed include Delavirdine (DLV), Efavirenz (EFV), Etravirine (ETR), Nevirapine (NVP), and Rilpivirine (RPV), with common adverse effects such as rash and hepatotoxicity. Next, accompanied by arrows pointing towards a blue sphere in the diagram, signify Nucleoside/Nucleotide Reverse Transcriptase Inhibitors (NRTI). Listed drugs include Abacavir (ABC), Didanosine (ddi), Emtricitabine (FTC), Lamivudine (3TC), Stavudine (d4T), Tenofovir (TDF), and Zidovudine (ZDV), with specific considerations for each drug, such as contraindications and preferred usage scenarios. The next column, with an arrow extending to the nucleus denote Integrase Inhibitors. Raltegravir (DRV), Elvitegravir (EVG), and Dolutegravir (RAL) are listed, along with a note on increased creatine kinase and their alternative name as integrase strand transfer inhibitors (INSTIs). Lastly, an arrow pointing towards a second blue sphere represent Protease Inhibitors. Atazanavir (ATV), Darunavir (DRV), Fosamprenavir (FPV), Indinavir (IDV), Lopinavir (LPV), Nelfinavir (NFV), Ritonavir (RTV), Saquinavir (SQV), and Tipranavir (TPV) are listed, along with common adverse effects and considerations for concurrent medication usage.
Figure 9.12: Mechanisms of resistance. Efflux pump (pumping drugs out of the cell): fluoroquinolones, aminoglycosides, tetracyclines, Beta-lactams, macrolides. Blocked penetration (not letting drugs into the cell): beta-lactams, tetracyclines, fluoroquinolones. Target modification (changing the target of the drug, such as ribosomes or DNA): fluoroquinolones, rifamycins, vancomycin, beta-lactams, macrolides, aminoglycosides. Inactivating enzyme (enzyme that breaks down the drug): beta-lactams, aminoglycosides, macrolides, rifamycins.
Figure References
Figure 9.1: Broad-spectrum antimicrobial use may lead to the development of a superinfection. Modification of work by Centers for Disease Control and Prevention. Public domain.
Figure 9.2: On this graph, t0 represents the time at which a drug dose is administered. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.3: There are several classes of antibacterial compounds that are typically classified based on their bacterial target. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.4: Penicillins, cephalosporins, monobactams, and carbapenems all contain a β-lactam ring, the site of attack by inactivating β-lactamase enzymes. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.5: The major classes of protein synthesis inhibitors target the 30S or 50S subunits of cytoplasmic ribosomes. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.6: Sulfonamides and trimethoprim are examples of antimetabolites that interfere in the bacterial synthesis of folic acid by blocking purine and pyrimidine biosynthesis, thus inhibiting bacterial growth. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.7: The predominant sterol found in human cells is cholesterol, whereas the predominant sterol found in fungi is ergosterol, making ergosterol a good target for antifungal drug development. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.8: Antifungal drugs target several different cell structures. Modification of work by (c) Maya and Rike. CC BY 3.0 Unported. https://commons.wikimedia.org/wiki/File:Cell_wall_structure_of_Fungi.png
Figure 9.9: Acyclovir is a structural analog of guanosine. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.10: Antiretroviral therapy (ART) is typically used for the treatment of HIV. The targets of drug classes currently in use are shown here. Modification of work (c) Thomas Splettstoesser. CC BY SA 4.0. https://commons.wikimedia.org/wiki/File:HI-virion-structure_en.svg
Figure 9.11: Targeting HIV replication. (c) Rice University. OpenStax Microbiology. CC BY 4.0. https://openstax.org/details/books/microbiology.
Figure 9.12: There are multiple strategies that microbes use to develop resistance to antimicrobial drugs. Modification of work (c) Gerard D Wright. CC BY 4.0.
Text References
- Wald-Dickler N, Holtom P, Spellberg B. Busting the Myth of "Static vs Cidal'': A Systemic Literature Review. Clin Infect Dis. 2018 Apr 17;66(9):1470-1474. doi: 10.1093/cid/cix1127. PMID: 29293890; PMCID: PMC5905615. ↵
- M.E. Falagas, D.E. Karageorgopoulos. “Adjustment of Dosing of Antimicrobial Agents for Bodyweight in Adults.” The Lancet 375 no. 9710 (2010):248–251. ↵
- B.D. Dickinson et al. “Drug Interactions between Oral Contraceptives and Antibiotics.” Obstetrics & Gynecology 98, no. 5 (2001):853–860. ↵
- Zhou W, Wang H, Yang Y, Chen ZS, Zou C, Zhang J. “Chloroquine against malaria, cancers and viral diseases.” Drug Discov Today. 2020 September. 16;25(11):2012–22. doi: 10.1016/j.drudis.2020.09.010. Epub ahead of print. PMID: 32947043; PMCID: PMC7492153. ↵
- B. Chu et al. “A Benzimidazole Derivative Exhibiting Antitumor Activity Blocks EGFR and HER2 Activity and Upregulates DR5 in Breast Cancer Cells.” Cell Death and Disease 6 (2015): e1686. ↵
- J.-X. Pan et al. “Niclosamide, An Old Anthelmintic Agent, Demonstrates Antitumor Activity by Blocking Multiple Signaling Pathways of Cancer Stem Cells.” Chinese Journal of Cancer 31 no. 4 (2012):178–184. ↵
- F. Imperi et al. “New Life for an Old Drug: The Anthelmintic Drug Niclosamide Inhibits Pseudomonas aeruginosa Quorum Sensing.” Antimicrobial Agents and Chemotherapy 57 no. 2 (2013):996-1005. ↵
- A. Jurgeit et al. “Niclosamide Is a Proton Carrier and Targets Acidic Endosomes with Broad Antiviral Effects.” PLoS Pathogens 8 no. 10 (2012): e1002976. ↵
- Park SK, Friedrich L, Yahya NA, Rohr CM, Chulkov EG, Maillard D, Rippmann F, Spangenberg T, Marchant JS. Mechanism of praziquantel action at a parasitic flatworm ion channel. Sci Transl Med. 2021 Dec 22;13(625):eabj5832. doi: 10.1126/scitranslmed.abj5832. Epub 2021 Dec 22. PMID: 34936384; PMCID: PMC8855674. ↵
- Centers for Disease Control and Prevention. “Antibiotic/Antimicrobial Resistance.” http://www.cdc.gov/drugresistance/index.html. Accessed June 2, 2016. ↵
- A.S. Kalokhe et al. “Multidrug-Resistant Tuberculosis Drug Susceptibility and Molecular Diagnostic Testing: A Review of the Literature. American Journal of the Medical Sciences 345 no. 2 (2013):143–148. ↵
- Centers for Disease Control and Prevention. “Methicillin-Resistant Staphylococcus aureus (MRSA): General Information About MRSA in the Community.” http://www.cdc.gov/mrsa/community/index.html. Accessed June 2, 2016. ↵
- Centers for Disease Control and Prevention. “Healthcare-Associated Infections (HIA): General Information about VISA/VRSA.” http://www.cdc.gov/HAI/organisms/visa_vrsa/visa_vrsa.html. Accessed June 2, 2016. ↵

{kind=link}
{kind=link}